TREATMENT
OF PRIMARY PULMONARY
HYPERTENSION - THE NEXT GENERATION
PRIMARY pulmonary hypertension predominantly
affects women, frequently in the prime of life, and usually
leads to death from right ventricular failure within a few years
after diagnosis. It is a vascular disease but is oddly confined
to the small pulmonary arterioles, where intimal fibrosis and
medial hypertrophy lead sequentially to vascular obstruction,
elevated pulmonary vascular resistance, pulmonary hypertension,
and right ventricular overload.
Coagulation at the endothelial surface contributes
to obstruction, and thromboembolism may occur as a secondary
event. The right ventricle compensates through hypertrophy,
and although it can sustain function at high pressures for months
to years, decompensation is ultimately manifested in reduced
cardiac output and the development of peripheral edema.
Many conditions and diseases lead to similar
pulmonary vascular lesions and clinical outcomes, including
the scleroderma spectrum of diseases, human immunodeficiency
virus infection, liver disease, and the use of certain anorectic
drugs.These illnesses, along with primary pulmonary hypertension,
are now classified as types of pulmonary arterial hypertension.
Primary pulmonary hypertension first came under coordinated
scientific scrutiny when the National Institutes of Health created
the national Primary Pulmonary Hypertension Patient Registry
in 1982, at a time when there was increasing optimism about
a role for vasodilator therapy. Although there had been multiple
previous reports of benefit from beta-agonists, alpha-blockers,
and hydralazine, these responses were usually not sustained,
and the relevant studies were not appropriately powered to detect
true effects.
The discovery that calcium-channel blockers
could cause a sustained reduction in pulmonary vascular resistance
in about 20 to 25 percent of previously untreated patients led
to aggressive approaches to short-term vasodilator testing and
long-term vasodilator therapy. Although not every patient with
acute vasodilatation has a durable response to therapy, this
feature carries a favorable prognosis, and many such patients
are treated with calcium-channel blockers alone. It has not
been proved that vasoconstriction is a pathogenetic mechanism
of primary pulmonary hypertension, but this possibility seems
logical and deserves continued study.
What can be done for the 75 percent of patients who do not have
a response to short-term vasodilator therapy? The discovery
that intravenous epoprostenol (prostacyclin) improved functional
capacity, not only in patients with a response to calcium-channel
blockers but also in those without a response, was followed
by evidence that it also improves survival among both types
of patients.This finding has led to widespread use of continuous
intravenous epoprostenol therapy in all patients without a response
to calcium-channel blockers and in most patients with New York
Heart Association class IV heart failure.
Beyond the activity of epoprostenol as a potent
vasodilator, its mechanisms of benefit are unclear, but they
may indude a positive inotropic effect, a small degree of systemic
vasodilatation, and antiplatelet effects, which theoretically
could reverse vascular damage.
Epoprostenol therapy by continuous infusion through a central
catheter is expensive - about $60,000 per year - as well as
technically demanding, and it has undesirable side effects.
It is widely recognized that simpler effective therapies are
needed. Prostacyclin analogues given by continuous subcutaneous
infusion, orally, or by intermittent aerosol are under development
as alternatives to the intravenous route. Subcutaneous treprostinil
was recently approved by the Food and Drug Administration for
further clinical trials. The prostacyclins act through an increase
in the level of the second messenger, intracellular cyclic AMP
(cAMP). Other vasodilators, including inhaled nitric oxide and
oral sildenafil, act by means of cyclic guanosine monophosphate
(cGMP).
Sildenafil increases the cGMP level by inhibiting
phosphodiesterase type 5, an enzyme that hydrolyzes cGMP. Clinical
studies are needed to test for potential additive effects of
simultaneous increases in cGMP and cAMP by combining the two
classes of drugs. Safe generation of nitric oxide in vivo might
be attained with the use of oral arginine or citrulline, substrates
for the generation of nitric oxide, with resultant cGMP levels
sustained by concomitant oral sildenafil.
Endothelin-1 is a potent endogenous peptide mediator that has
a role in pulmonary arterial hypertension. It is unclear whether
it has a primary pathogenetic role or whether it is a secondary
mediator that perpetuates disease. Plasma endothelin levels
are increased in patients with primary pulmonary hypertension,
and endothelin is released in increased amounts in the blood
traversing the lung. Endothelin is released by endotheial cells
as big endothelin, which is cleaved to pro-endothelin, which,
in turn, is converted to endothelin-1 (in systemic and lung
vessels) or endothelin-2 (in kidney and gut). Endothelin-1 acts
on two receptors - endothein-A receptors and endothein-B receptors.
Activation of endothelin-B receptors causes the production of
nitric oxide and vasodilatation, and activation of endothelin-A
receptors results in vasoconstriction and smooth-muscle growth.
The ideal endothein-receptor antagonist is likely to be specific
for endothelin-A.
A study using bosentan, a nonspecific endothelin receptor antagonist,
to treat pulmonary hypertension is reported in this issue of
the Journal ( N Engl Med,Vol.346,No.12,March21,2002).
Bosentan(tracleer) had small but measurable beneficial effects
in a double-blind, placebo-controlled trial involving 213 patients.
The duration of this trial was 16 weeks, which is not sufficient
to test for a difference in mortality, but its results suggest
that endothelin-receptor blockade has a therapeutic role in
some patients with pulmonary arterial hypertension. The effect
of bosentan appeared to be limited in most patients, and there
was an unacceptable incidence of abnormal hepatic function at
the higher dose(elevation of liver aminotransferases,sgot and
sgpt). Because short-term vasodilator testing was not performed
as part of the study, it is not known whether the patients with
the best response to the drug were the same patients who might
have had a response to other vasodilators. One cannot conclude
from this study that bosentan should be the primary drug for
the treatment of primary pulmonary hypertension or of other
causes of pulmonary arterial hypertension.
Follow-up studies are needed to determine the
durability of the effect, whether there are differences in survival,
what types of complications occur, and whether subgroups of
patients have different responses to the drug. It would be useful
to measure endothelin levels and to determine whether there
are correlations between these levels and clinical effects.
Studies should be designed to test whether combining endothelin-receptor
antagonists with either inhibitors of phosphodiesterase type
5 or inducers of cAMP results in greater functional improvement
than does either class of drug alone.
No current therapies appear to affect the pathogenesis of pulmonary
vascular obstructive disease directly. In rare cases, patients
receiving epoprostenol have had such dramatic responses that
the dose has been reduced, and cessation of drug therapy has
been attempted in a few patients, although the outcomes have
not been published.
The recent discovery that the transforming growth
factor beta(TGF-beta) superfamily of receptors is involved in
the pathogenesis of pulmonary hypertension should lead over
the course of the next several years to specific therapies aimed
at the origin of the disease. The evidence suggesting the involvement
of TGF-beta receptors is compelling. About half of studied patients
with familial primary pulmonary hypertension have mutations
in exons of the bone morphogenetic protein receptor II gene
(BMPR2), and the majority of others have genetic linkage to
areas of chromosome 2 near BMPR2, perhaps in a promoter or upstream
regulator or perhaps in intronic DNA. In addition, about 25
percent of patients with sporadic primary pulmonary hypertension
have been found to have mutations in BMPR2.
Mutations in the gene for activin-receptor-like
kinase 1 (ALK1), another receptor in the TGF-beta family, are
responsible for pulmonary hypertension in at least some patients
with hereditary hemorrhagic telangiectasia. Clusters of endotheial
cells carrying somatic TGF-beta 2-receptor mutations are found
in plexiform lesions in the pulmonary arterioles of patients
with sporadic primary pulmonary hypertension. Studies of these
receptor abnormalities in transfected cells, cell cultures from
patients' tissues, and transgenic mice are under way, and insights
into the relevant mechanisms will certainly emerge during the
next several years. Other promising areas of research are potassium-channel
function and drugs that interrupt the cycle of growth and repair
in diseased pulmonary vessels.
Therapy for primary pulmonary hypertension has progressed from
calcium-channel blockers to prostacyclin and now includes adjunctive
therapy with bosentan and, in some patients, sildenafil. Combination
therapies should be tested in the next generation of studies.
It now seems conceivable that the continuous intravenous administration
of epoprostenol through a central catheter will soon be history.
A better understanding of pathogenesis is at hand because the
genes associated with many cases of primary pulmonary hypertension
have been identified, but the development of therapies based
on this knowledge awaits further insights.
JOHN H. NEWMAN, M.D.
Vanderbilt University School of Medicine
Nashville, TN 37232
Newman,J.H.,N Engl Med,Vol.346,No.12,March21,2002
Pp.933-935.
New Approaches to Pulmonary Hypertension
Careful evaluation will often reveal secondary--and perhaps
reversible--factors contributing to pulmonary hypertension.
For primary disease, there are now a variety of treatments,
ranging from calcium channel blockers to lung transplantation.
Until recently, therapeutic options in pulmonary hypertension
were limited. Disease caused or complicated by cardiac, respiratory,
or embolic factors could be treated by correction of those factors.
The pathophysiology of primary disease was poorly understood,
and patients could be offered little encouragement with regard
to prognosis or the likelihood of a favorable response to available
therapies. That picture changed with the discovery of a likely
mechanism for primary pulmonary hypertension (PPH): deficient
release of vasodilator mediators from pulmonary endothelium.
The discovery has led to an expanded palette of possible treatments
that can offer patients a reasonable expectation of stabilization
or improvement, and in some cases prolongation of survival.
The traditional classification of pulmonary hypertension into
primary and secondary categories is unsatisfactory for a number
of reasons. First, PPH is a clinical diagnosis. Second, the
distinction between primary and secondary disease depends on
how thoroughly one excludes secondary factors. For example,
routine use of studies usually not performed, such as pulmonary
angiography, might identify more cases of secondary disease.
http://www.emedicine.com/MED/topic1962.htm,Ronald J.Oudiz,MD.,10/23/02)
Pulmonary angiography continues to be the gold standard for
defining the pulmonary vascular anatomy and is performed to
identify whether chronic thromboembolic obstruction is present,
to determine its location and surgical accessibility, and to
rule out other diagnostic possibilities. Despite concerns regarding
the safety of performing pulmonary angiography in patients with
pulmonary hypertension, with careful monitoring and modification
of standard angiographic procedures, pulmonary angiography can
be performed safely even in patients with severe pulmonary hypertension.
Biplane imaging is preferred, offering the advantage of lateral
views that provide greater anatomic detail compared with the
overlapped and obscured vessel images often seen in the anterior-posterior
view. Interpre-tation of these angiograms can be difficult in
large measure because the appearance of chronic thromboemboli
bears little resemblance to the well-defined, intraluminal filling
defects of acute pulmonary embolism. Maturation and organization
of clot results in vessel retraction and partial recanalization
resulting in several angiographic patterns suggestive of chronic
thromboembolic disease: (1) pouch defects; (2) pulmonary artery
webs or bands; (3) intimal irregularities; (4) abrupt narrowing
of major pulmonary vessels; and (5) obstruction of main, lobar,
or segmental pulmonary arteries, frequently at their point of
origin (Figure 14).
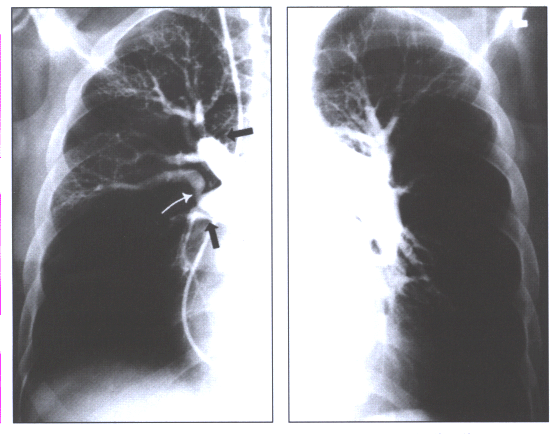 Fig.14:Angiographic
findings of chronic thromboembolic disease:pouches in the right
upper lobe and interlobar artery (black arrows),a band with
post-stenotic dilatation(white arrow),and rapid tapering of
the left descending pulmonary artery.
Fig.14:Angiographic
findings of chronic thromboembolic disease:pouches in the right
upper lobe and interlobar artery (black arrows),a band with
post-stenotic dilatation(white arrow),and rapid tapering of
the left descending pulmonary artery.
However, competing diagnoses exhibit angiographic findings
similar to those encountered with chronic thromboembolic disease.
For instance, areas of focal vessel narrowing, or "bands,"
can be seen as a feature of congenital stenosis of the pulmonary
arteries as well as of medium- or large-vessel arteritis. Total
obstruction or abrupt narrowing of the central pulmonary arteries
can be a feature of an intravascular process such as pulmonary
vascular tumors or extravascular compression from lung carcinoma,
hilar or mediastinal adenopathy, or mediastinal fibrosis. Since
chronic thromboembolic disease is usually bilateral, the presence
of unilateral central pulmonary artery obstuction should always
prompt consideration of one of these rival diagnoses.
In approximately 25% of patients evaluated at the University
of California, San Diego, pulmonary angioscopy is used to supplement
the information obtained from pulmonary angiography. The pulmonary
angioscope is a diagnostic fiberoptic device that was developed
to visualize the intima of central pulmonary arteries. It is
inserted through a vascular sheath inserted in a central vein
and passed through the right heart into the pulmonary artery
under fluoroscopic guidance. Inflation of a latex balloon affixed
to the tip of the angioscope results in obstruction of blood
flow in the artery and permits visualization of the arterial
intima. The most useful role for pulmonary angioscopy is in
identifying operative candidates whose angiographic findings
suggest limited disease (K.M.Kerr,MD,P.F.Fedullo,MD,and W.R.Auger
Chronic thromboembolic Pulmonary Hypertension:When to Suspect
It,When to Refer fro Surgery,Advances in Pulmonary Hypertension,J.Pulmonary
Hypertension,V2,No1,pp4-10).
Third, the distinction depends on whether any secondary factors
that may be present are considered sufficient to explain the
condition (e.g., PPH associated with portal hypertension).
(PPH is associated with portal hypertension. This suggests
that those patients with shunting of splanchnic blood, with
or without liver disease, have a higher risk of developing PPH.
Also, substances in the splanchnic circulation may contribute
to the development of pulmonary hypertension, and the liver
serves to detoxify the body of these substances. More research
is necessary to better understand this relationship.
http://www.emedicine.com/MED/topic1962.htm,Ronald J.Oudiz,MD.,10/23/02)
Terminology is of more than academic concern, because third-party
payers now reimburse for certain pharmacotherapies based on
whether the diagnosis is primary or secondary disease. Finally,
the current classification scheme makes no allowance for mixed
forms of pulmonary hypertension, as when a secondary factor
is identifiable but offers only a partial explanation.
Pathophysiology
Several physiologic factors contribute to pulmonary artery (PA)
pressure. Included are PA wedge pressure, cardiac output, and
pulmonary vascular resistance, increases in any of which may
result in pulmonary hypertension(PathophysiogyPulmonart hypertension-fig8).
For example, if PA wedge pressure rises, PA pressure must also
rise in order to maintain blood flow. Elevation of PA wedge
pressure, such as from left ventricular failure, is a very common
cause of pulmonary hypertension seen in acute care hospitals.
During exercise, cardiac output may increase as much as three-
to fivefold, particularly in conditioned athletes, and the increased
pulmonary blood flow raises PA pressure. In healthy persons,
the increase in pressure is much less than the increase in blood
flow because the vasculature can accommodate large increases
in flow via distention and recruitment. Moreover, exercise-related
increases in pulmonary blood flow are temporary. In contrast,
sustained increases--as with atrial or ventricular septal defects--can
lead to secondary changes in pulmonary arteries, including thickening
of vascular walls. Pulmonary vascular resistance increases,
and in combination with the increased blood flow, PA pressure
rises substantially.

Pathophysiology Pulmonary hypertension-fig8
Increased blood viscosity and diminished vessel radius are
the main contributors to increased pulmonary vascular resistance.
In the pulmonary circuit, increases in viscosity are almost
entirely attributable to increases in red cell concentration.
If hematocrit rises from 40% to 60%, vascular resistance doubles.
Most patients with increased pulmonary vascular resistance have
a diminished effective vessel radius. Causes include vessel
destruction, as may occur in emphysema or pulmonary fibrosis;
resection, as from lung surgery; or obstruction, as with pulmonary
embolism. As a rule, those processes do not cause significant
elevation of PA pressure unless 50% to 70% of the pulmonary
circulation has been occluded.
The most common cause of increased pulmonary vascular resistance
associated with chronic respiratory disease is hypoxia, which
causes vasoconstriction, thickening of the vascular media (remodeling),
and polycythemia. Despite intensive study during the past 50
years, the mechanisms responsible for hypoxic pulmonary vasoconstriction
remain undefined. Research in the past decade, however, has
suggested roles for potassium channels, nitric oxide, and endothelin,
among other factors (Figure 1).

Figure1: Pathophysiology of Pulmonary Hypertension
Table 1. Symptoms and Signs
of Pulmonary Hypertension |
|
| SYMPTOMS |
|
| |
Exertional dyspnea
Exertional chest pain or lightheadedness
Chest pounding during exertion
Exertional syncope
Cough
Hemoptysis |
| SIGNS |
|
Cardiac
Examination |
Increased intensity of P2
Right ventricular impulse or heave
Murmurs of tricuspid regurgitation
or pulmonic insufficiency
Right-sided gallops (Carvallo's sign)
Neck vein distention |
| Extremities |
Right ventricular impulse or heave
Raynaud's phenomenon |
Primary Subtypes
PPH includes a variety of pathologic conditions (Figure 2).
The most common are primary plexogenic pulmonary hypertension,
thromboembolic pulmonary hypertension, and pulmonary venoocclusive
disease. In the absence of open-lung biopsy, each is usually
identified postmortem. Other entities that may occasionally
present as PPH include intravenous drug use, tumor emboli, occult
interstitial lung disease, occult chronic hypoxia, and schistosomiasis.

Fig. 2 Pathology of various types of pulmonary hypertension
Primary plexogenic arteriopathy is characterized pathologically
by the plexiform lesion, a disorganized whorl of capillarylike
vessels(see pulmonaryHTN-fig3 below). Some of these lesions
consist of a monoclonal proliferation of endothelial cells.
The plexiform lesion may be seen in a variety of forms of severe
pulmonary hypertension.

Pulmonary hypertension lesion-fig3-jpg
Primary plexogenic arteriopathy has an incidence of one to
two per million and a female preponderance of 1.7:1. Age at
diagnosis typically ranges from 20 to 50 years. Most patients
are otherwise healthy young women. The disorder appears to have
a genetic component: 7% of cases are familial. Indeed, the mutation
involved has been traced to a specific locus on chromosome 2.
Plexogenic arteriopathy has also been associated with toxin
ingestion, pregnancy, cirrhosis, HIV infection, connective tissue
disease, and use of appetite suppressants.
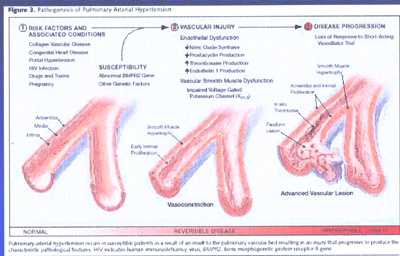
Pathogenesis of Pulmonary Arterial Hypertension(FIG4)
It occurs in susceptible patients as a result of an insult
to the pulmonary vascular bed resulting in an injury that progresses
to produce the characteristic pathogologoic features
Recent studies on the etiology of the disease suggest that
pulmonary endothelial cell injury leads to an imbalance of vasoconstrictor
over vasodilator agents and to the release of growth factors
that may promote vessel-wall thickening. The combination raises
pulmonary vascular resistance, leading to progressive pulmonary
hypertension(see fig5,fig6 and fig7 below).
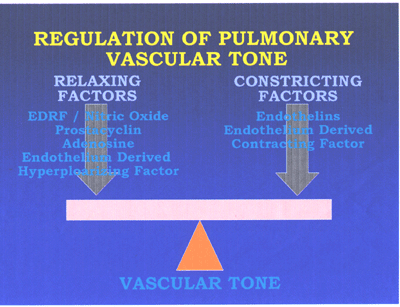
RegPulmVasctone-fig5

Fig6. Pathophysiology of pulmonary hypertension
Pathophysiology of pulmonary hypertension-fig7
Thromboembolic Pulmonary Hypertension. Repeated episodes of
embolism that do not resolve may eventually occlude enough pulmonary
vasculature to cause hypertension. Pathologically, these cases
are marked by such characteristic lesions as eccentric intimal
thickening and webs and septa within the arterial lumen.
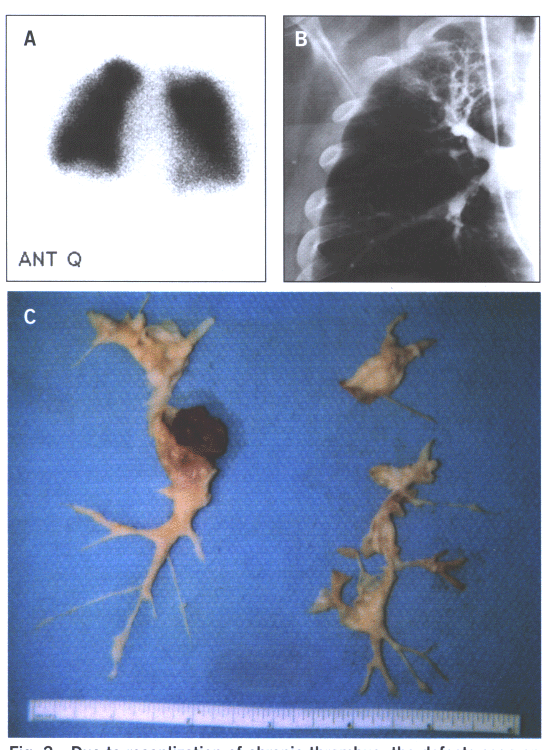
Fig.15: Due to recanalization of chronic thrombus,the defects
seen on the perfusion scan 15A grossly understate the degree
of obstruction seen on the pulmonary angiogram (15B) and the
findings at the time of surgery(15C).
Thromboembolic pulmonary hypertension has two forms: macrothromboembolic(fig.15),
which is usually considered a type of secondary pulmonary hypertension
and consists of large clots that occlude central vessels; and
microthromboembolic, in which distal thrombi occlude many smaller
vessels. The macro form usually responds to thromboendarterectomy
and should always be sought in the evaluation of patients with
suspected PPH. The micro form may be related to in situ thrombosis
and appears to overlap clinically with primary plexogenic arteriopathy(fig.16);
both patterns have been described in families with pulmonary
hypertension.
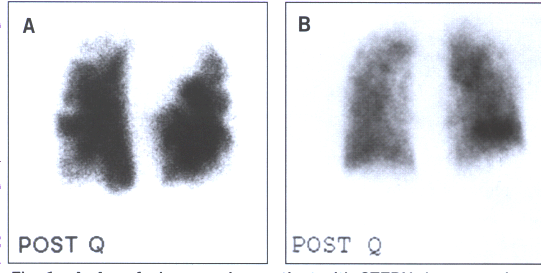
Fig.16: A.A perfusion scan in a patient with chronic thromboembolic
pumonary hypertension demonstrating multiple,bilateral,segmental
perfusion defects. B.A patient with PPH with a "mottled"
perfusion scan without any segmental perfusion defects.
Pulmonary Venoocclusive Disease. This less common entity is
caused by intimal thickening of pulmonary venules. It is seen
mainly in younger men and may respond to steroids.
Associated Conditions
Pulmonary hypertension is commonly associated with connective
tissue diseases, including systemic lupus erythematosus, mixed
connective tissue disease, and progressive systemic sclerosis;
in the limited form of progressive systemic sclerosis, at least
mild pulmonary hypertension has been reported in up to 50% of
patients. Underlying pulmonary fibrosis may also contribute
to the hypertension. Raynaud's phenomenon is noted in up to
10% of cases of PPH and in most patients with connective tissue
disease-associated pulmonary hypertension.
Appetite suppressants gained notoriety as a cause of pulmonary
hypertension in 1997, when the Food and Drug Administration
(FDA) withdrew the serotonin uptake inhibitors fenfluramine
and dexfenfluramine from the U.S. market. (Fenfluramine had
often been used together with the sympathomimetic anorectic
phentermine, a combination known as fen-phen.) Fenfluramine
and dexfenfluramine were associated with up to a 23-fold increase
in the incidence of PPH, possibly due to pulmonary vasoconstriction
and remodeling from increased circulating serotonin. As would
be expected, the incidence of PPH appears to be falling since
the ban of those drugs.
Portopulmonary hypertension--the combination of portal hypertension
and PPH--occurs in at least 1% of patients with cirrhosis. One
theory posits that vasoconstriction and remodeling result from
the accumulation of circulating mediators that are metabolized
by the healthy liver. Although specific mediators have yet to
be identified, this theory is supported by the observation that
some patients with portopulmonary hypertension experience reversal
of their disease after liver transplantation.
Pulmonary hypertension occurs in at least 1% of HIV-positive
patients and can develop at any stage of the illness. Pathologic
findings are similar to those of PPH, including the occasional
patient with venoocclusive disease. Foreign-body emboli may
contribute to this problem in patients with a history of intravenous
drug abuse, and portal hypertension may be present in those
patients with associated hepatitis C. The etiologic role of
HIV itself in pulmonary hypertension remains unknown.
Clinical Evaluation
Dyspnea on exertion and fatigue are the cardinal symptoms of
pulmonary hypertension ( see Table 1 above). More severe cases
manifest with exertional chest pain or dizziness. Exertional
syncope is a worrisome development. Occasional patients experience
hemoptysis, cough, or hoarseness; the last is thought to be
related to the pressure of dilated pulmonary arteries on the
left recurrent laryngeal nerve.
Signs of pulmonary hypertension derive from compensatory changes
in the right ventricle. The earliest and most sensitive manifestation
is an increase in the intensity of the second heart sound in
the pulmonic area (P2). To elicit this sign, one compares the
intensity of the sound in the pulmonic area at the left upper
sternal border with that in the aortic area at the right upper
sternal border. Normally, A2 is more intense than P2. With an
increase in pulmonary artery pressure, however, the pulmonic
valve shuts more vigorously and P2 becomes louder.
As pulmonary hypertension advances, a right ventricular impulse
may be palpable at the left lower sternal border. With marked
right ventricular dilation, a heave becomes perceptible. Right-sided
gallops and tricuspid regurgitant murmurs are also common findings.
Neck-vein distention with a prominent V-wave, a murmur of pulmonic
insufficiency, and pronounced lower-extremity edema are all
signs of advanced pulmonary hypertension, which is often associated
with a thready systemic pulse.
Laboratory Findings
A 12-lead ECG and chest x-rays are among the routine tests for
suspected pulmonary hypertension (Table 2). The ECG may show
right ventricular hypertrophy or strain or right atrial enlargement.
ECG findings include right axis deviation (QRS axis >110);
P-pulmonale with a P wave height greater than 2.4 mm in lead
II; right bundle branch block; and an R height greater than
5 mm or R/S ratio greater than 1 in lead V1. Down-sloping ST
depressions in the anterior precordial leads are seen with right
ventricular strain. The chest film may show dilation of the
right descending pulmonary artery (>16 mm in diameter for
women and >18 mm for men) and "pruning" of peripheral
vessels. Filling of the retrosternal air space on the lateral
view indicates right ventricular enlargement.
|
Table 2. Diagnostic Evaluation
of Pulmonary Hypertension
ROUTINE TESTS
CBC, sedimentation rate
Liver function tests including hepatitis screen
Thyroid function tests
Connective tissue screen with anticardiolipin antibody
HIV antibody test
Electrocardiogram
Chest x-ray
Ventilation/perfusion lung scan
PULMONARY FUNCTION
Spirogram, lung volume, and Dlco
Arterial blood gas determinations
Exercise oximetry
FUNCTIONAL STATUS
6-Minute walk test*
New York Heart Association Classification
NONINVASIVE HEMODYNAMIC EVALUATION
Transthoracic echocardiogram with Doppler
Special circumstances: transesophageal or stress echocardiogram,
radionuclide or MRI scan, CT scan
SLEEP EVALUATION
Polysomnogram if features suggest obstructive sleep apnea
INVASIVE HEMODYNAMIC EVALUATION
Right-heart catheterization
02 saturation step-up
Vasodilator trial
Left-heart catheterization
If left heart function in doubt or coronary artery disease
is suggested
OPEN LUNG BIOPSY
If steroid-responsive disease is suspected (i.e., pulmonary
fibrosis, vasculitis, pulmonary venoocclusive disease)
*Maximal stress testing discouraged
|
The most useful noninvasive test is transthoracic echocardiography,
which should include Doppler estimation of the PA pressure,
based on the velocity of the tricuspid or pulmonic regurgitant
jet(Figs.9,10,11,12). Pulmonary hypertension is defined as a
mean PA pressure exceeding 25 mm Hg at rest (30 mm Hg during
exercise). Mean pressures of 26 to 35 mm Hg are considered mild
elevations; those of 36 to 45 mm Hg moderate; and greater than
45 mm Hg severe.
Echocardiography is also helpful in excluding mitral valve
lesions or myxomas that may contribute to pulmonary hypertension.
With advanced disease, right ventricular size and function are
usually abnormal; paradoxic septal motion and abnormal pulmonic
valve motion may be apparent by echocardiography.
However, echocardiographic determinations of right ventricular
or pulmonary artery systolic pressure are merely estimates.
Although in general they correlate very well with invasive measurements
of right ventricular pressures, an individual estimate may be
inaccurate. In particular, echocardiography has only limited
ability to differentiate between mild pulmonary hypertension
and normal pulmonary hemodynamics.
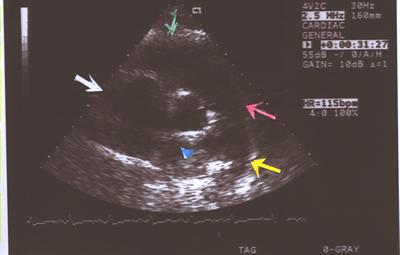
Fig.9.Two- dimensional Echocardiogram showing an enlarged main
pulmonary artery(red arrow),right pulmonary artery(blue arrow
head),left pulmonary artery(yellow arrow),right ventricle(green
arrow) and right atrium(white arrow) in a short axis view in
a case of primary pulmonary hypertension(PPHTN) with a Doppler
right ventricular systolic pressure of 100mm.Hg.

Normal heart, branch
pulmonary arteries
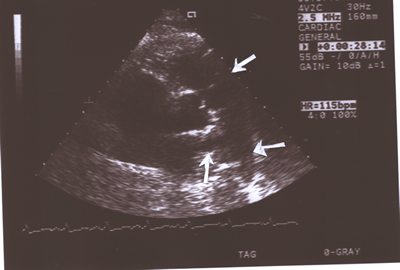
Fig.10:Same as Fig.9 above but showing the pulmonary valve
( upper white arrow)better (short axis,parasternal view) and
the right( lower upright arrow)and left(lower horizontal arrow)
pulmonary arteries.
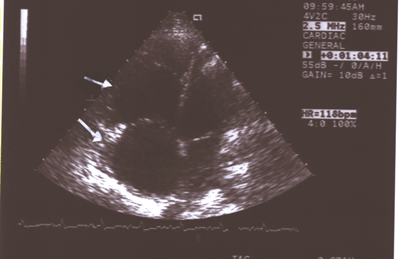
Fig.11:Same as Fig.9 showing the enlarged right atrium(larger
arrow)and right ventricle(smaller arrow)compared to the much
smaller left atrium and ventricle(4-chamber long axis view).
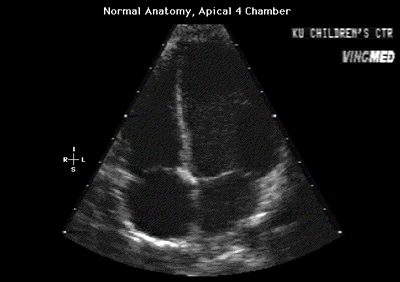
Fig.11a:Normal anatomy,apical 4 chamber view to compare with
Fig.11 above with the enlarged right heart.
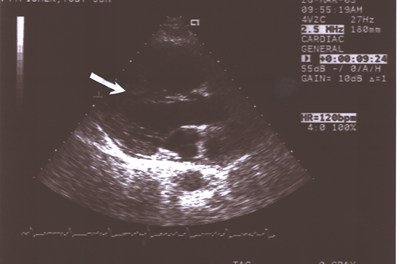
Fig.12: Same as Fig.9 showing the enlarged right atrium and
ventricle separated by the straightened interventricular septum(ISV,arrow),which
was paradoxical in motion ,compared to the much smaller left
atrium and ventricle(long axis parasternal view).
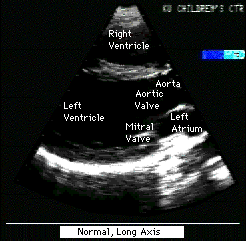
Fig.12a:Normal,parasternal,long axis view of heart to compare
with Fig.12 above.

Fig.13:ECG from above patient with PPHTN showing right atrial
enlargement(tall peaked P waves),minus14 degree QRS axis and
diffuse T wave inversions,mainly in V1-5 leads,with clockwise
rotation of the heart,possibly due to right heart strain.A left
heart catheterization showed normal coronary arteries ,mild
mitral regurgitation and a reduced LV ejection fraction.
Further testing may be indicated in some cases. Transesophageal
echocardiography may be used to detect intracardiac shunts or
better visualize valvular structures. A stress echocardiogram
or radionuclide studies may be helpful when a more complete
evaluation of right ventricular function is desired.
When pulmonary hypertension is documented, a series of additional
tests should be performed to exclude secondary factors and to
seek associated conditions. These include a hemogram, pulmonary
function tests, exercise oximetry or an arterial blood gas determination,
a radionuclide lung scan, a connective tissue screen including
a cardiolipin antibody test, liver function studies, thyroid
function tests, and HIV testing. If the lung scan is suspicious
for pulmonary embolism, a pulmonary arteriogram should be performed.
A polysomnogram is indicated if the clinical presentation suggests
obstructive sleep apnea.
The six-minute walk test is useful for determining prognosis
and for establishing a baseline to follow response to therapy.
This test requires that the patient walk as far as possible
on a measured course for six minutes while oxygen saturation
is monitored. Patients who can walk less than 250 to 300 meters
are considered to have severe exercise limitation. Some clinicians
perform computed tomographic angiography instead, but the utility
of this approach has not been established.
If no secondary factors can be identified that explain the
pulmonary hypertension, or if an evaluation of vascular reactivity
is desired, right heart catheterization is indicated. This test
confirms the clinical diagnosis of primary disease (by excluding
congenital left-to-right shunt or left ventricular dysfunction)
and provides an accurate assessment of PA pressure. It also
permits testing of responses to a variety of potential pharmacotherapies.
The acute vasodilator response is now an important part of the
diagnostic evaluation because it aids in the selection of a
vasodilator regimen and imparts prognostic information.
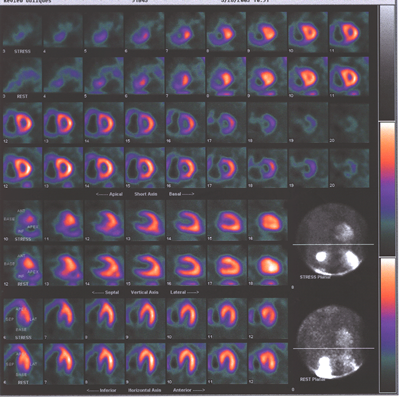
Fig.13,Myoview-a: nuclear scan showing right heart enlargement(dark
shadow next to pink shadow which represents left ventricle),worse
after stress,no perfusion defects.
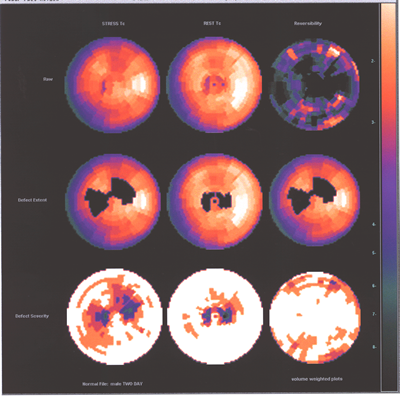
Fig.13,Myoview-b:Same patient as above with no perfusion defect
post stress.
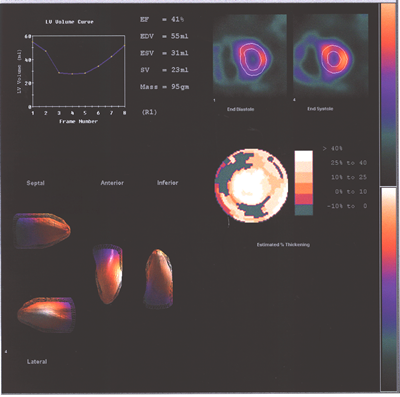
Fig.13,Myoview-c:Same patient as above with a reduced LV ejection
fraction at rest and the right ventricle enlargement increasing
with stress.
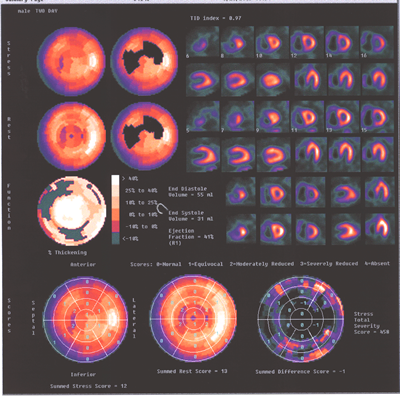
Fig.13,Myoview-d:Same patient as above.showing again the marked
difference inthe sizes ot the two ventricles.
Medical Therapy
Targeting Secondary Factors. Treatment of pulmonary hypertension
begins with attempts to reverse any identified contributing
factors, which often bring substantial clinical improvement.
For instance, pulmonary hypertension associated with severe
polycythemia (hematocrit >56%) may respond to phlebotomy,
and that associated with obstructive sleep apnea may improve
with nasal continuous positive airway pressure. Patients with
reversible airway obstruction should receive bronchodilator
therapy; those with marked right ventricular dysfunction and
edema often respond symptomatically to salt restriction, diuretics,
and digoxin.
Most important, the potential therapeutic and survival benefits
of oxygen supplementation in patients with chronic hypoxemia
should never be overlooked.
Anticoagulation. Warfarin is widely recommended for patients
with severe primary or secondary pulmonary hypertension to prevent
in situ thrombosis formation and venous thromboembolism. Most
pulmonologists treat to an International Normalized Ratio of
1.5 to 2.5.
Vasodilator Therapy. The goal of vasodilator therapy is to
reduce right ventricular overload by relaxing pulmonary vessels
and, ideally, reversing vessel remodeling. Until recently, calcium
channel blockers were the only vasodilator class associated
with improved survival in pulmonary hypertension. In an uncontrolled
prospective trial, for example, S. Rich and B. H. Brundage demonstrated
that acute vasodilation (>25% drop in pulmonary vascular
resistance) in response to nifedipine or diltiazem during right
heart catheterization correlated with excellent long-term survival
(approaching 90% at five years). Only about 20% to 25% of patients
respond favorably to calcium channel blockers, however.
In the early 1990s, researchers observed that patients with
pulmonary hypertension are relatively deficient in the release
of prostacyclin, a vasodilatory prostaglandin. This led to the
most important pharmacotherapeutic development in the field
of the past decade: continuous intravenous infusion of synthetic
prostacyclin (epoprostenol). Studies have confirmed a significant
improvement in both clinical manifestations and long-term survival
with this approach, and it is now considered the therapy of
choice for selected patients with severe primary disease (with
or without connective tissue disease) unresponsive to calcium
channel blockers.
Candidates for epoprostenol should be selected carefully because
infusion requires a permanently implanted central venous catheter,
and the patient must reconstitute the drug daily to maintain
a continuous infusion. Meticulous care is necessary to avoid
catheter infection, with its associated morbidity and mortality.
As tachyphylaxis develops, the dose of epoprostenol is gradually
adjusted upward. Titration involves balancing manifestations
of drug excess (headache, jaw ache, nausea, leg or foot pain,
and diarrhea) against those of drug deficiency (increased exertional
dyspnea). Sudden cessation of long-term infusion is poorly tolerated,
causing an abrupt return of dyspnea. At higher doses, the cost
of treatment may exceed $100,000 annually. Because of the risks,
expense, and inconveniences of continuous intravenous infusion,
other modes of prostacyclin administration are under evaluation.
Another endogenous vasodilator, nitric oxide, shows promise
as a long-term therapy for pulmonary hypertension, but only
preliminary trials have been reported. Inhaled nitric oxide
was recently approved by the FDA for PPH of the newborn; off-label
use as long-term therapy for adults is prohibitively expensive,
and no pharmaceutical companies are currently supporting trials
of it. Other promising new agents that have not yet been tested
in controlled trials include thromboxane and endothelin receptor
antagonists. Gene therapies that would promote synthesis of
nitric oxide, prostaglandin, or natriuretic peptide are being
tested in animal models.
Vasodilator therapy is best initiated at a center specializing
in the management of pulmonary hypertension (Figure 3). During
right-heart catheterization, acute vasoreactivity is tested
using intravenous adenosine or epoprostenol or inhaled nitric
oxide. If pulmonary vascular resistance drops by more than 25%,
a trial of calcium channel blockers is indicated. Most clinicians
use nifedipine, diltiazem, or amlodipine rather than verapamil
because of concerns about verapamil's negative inotropic effects.
Some advocate high-dose therapy, increasing nifedipine doses
to 480 mg or diltiazem to 720 mg daily if necessary.

Figure 3: Approach to Vasodilator Therapy for Pulmonary Hypertension
If pulmonary vascular resistance drops below 25%, patients
with New York Heart Association class II functional capacity
may still receive a trial of calcium channel blocker therapy,
but those with class III or IV symptoms should be considered
for intravenous epoprostenol or other investigational therapy
and referral for lung transplantation. Because of its expense,
epoprostenol therapy requires prior approval by a third-party
payer.
Surgery
Surgical therapy for severe disease is appropriate in special
circumstances. Thromboendarterectomy is highly effective for
patients with chronic thromboembolism who have a central clot
that fails to resolve after at least six months of anticoagulant
therapy. Careful dissection and removal of the clot leads to
significant reductions in PA pressure and vascular resistance
and a consistent improvement in functional status. Few centers
have extensive experience with this procedure, however, with
the exception of the University of California, San Diego.
Surgical Selection
Pulmonary endarterectomy is considered in patients who are symptomatic
and have evidence of hemodynamic or ventilatory impairment at
rest or with exercise. Patients undergoing surgery usually exhibit
a preoperative pulmonary vascular resistance greater than 300
dynes/sec/cm-5, typically in the range of 8001000 dynes/sec/cm-5.
For those with milder pulmonary hypertension, the decision to
operate is based on individual circumstances. Some with mild
elevation in pulmonary pressures at rest may develop a significant
rise in pressure with exertion. While not yet substantiated,
it is suspected these elevated pressures over a prolonged period
of time contribute to the development of small-vessel arteriopathy
in the patent vascular bed. Some patients may elect to undergo
surgery at this early stage of disease because of dissatisfaction
with their exercise limitation or concerns about clinical deterioration
in the future. Those who choose not to pursue surgical intervention
at this stage of their disease require close monitoring for
progression of pulmonary hypertension. Thromboendarterectomy
is also considered in patients with normal or nearly normal
hemodynamics with significant involvement of one pulmonary artery,
those with lifestyles that involve vigorous activity (eg athletes),
and those who live at higher altitude. Dyspnea in these patients
is a function of elevated dead space and minute ventilation
requirements and suboptimal cardiac output with higher level
exercise.
Operability is determined by the location and extent of proximal
thromboemboli. The experience of the surgical team will determine
what is considered surgically accessible. Thrombi must involve
the main, lobar, or proximal segmental arteries; disease originating
more distally is not accessible with current endarterectomy
techniques. Crucial to determining surgical candidacy and predicting
operative outcome is determining whether the amount of surgically
accessible thrombus is compatible with the degree of hemodynamic
impairment. This is particularly true in patients with severe
preoperative pulmonary hypertension and right ventricular dysfunction.
Failure to significantly reduce the pulmonary vascular resistance
with endarterectomy, usually a result of secondary small-vessel
arteriopathy, is associated with a greater perioperative mortality
rate and a worse long-term outcome."
The assessment of comorbid conditions is the next step in preoperative
surgical evaluation. Severe left ventricular dysfunction is
the only absolute contraindication to pulmonary thromboendarterectomy.
Advanced age, severe right ventricular dysfunction, and other
significant comorbid illnesses increase the perioperative morbity
and mortality, but these do not preclude surgical consideration.
Pediatric patients and octogenarians, as well as those with
complex coexistent disease have successfully undergone the surgical
procedure. Patients at risk for coronary atherosclerotic disease
should undergo coronary angiography preoperatively and coronary
artery bypass grafting or valve replacement can be performed
at the time of endarterectomy.
Referring for Pulmonary Endarterectomy
Since surgery has the potential to substantially improve symptoms
and pulmonary hemodynamics and the long-term outcome is poor
in medically treated patients, pulmonary thromboendarterectomy
should be considered in any patient once the diagnosis of CTEPH
is made. Prior to surgery, most patients are in New York Heart
Association functional class III or IV but postoperatively are
in class I or II and able to resume normal activities. Approximately
2000 endarterectomy procedures have been performed worldwide,
with roughly 1500 of them done at one center. In a review of
surgical series published since 1996, perioperative mortality
rates ranged from 5% to 24%, with significant variation in hemodynamic
improvement reported.' Given the high risk of pulmonary endarterectomy,
patients should be referred to centers that are able to provide
a multidisciplinary team with experience in the details of the
evaluation and treatment of chronic thromboembolic disease.
Since perioperative morbidity and mortality are significantly
influenced by the degree of right ventricular dysfunction and
the presence of secondary small-vessel vasculopathy, surgical
intervention is best pursued sooner in the disease process rather
than waiting until the patient suffers from significant clinical
and hemodynamic impairment.
Patients who are not candidates for thromboendarterectomy, and
those who suffer from significant residual pulmonary hypertension
following surgery, should be considered for lung transplantation.
Long-term treatment with epoprostenol may also be of benefit
in selected patients.30 The long-term efficacy of prostacyclin
analogs, endothelin-receptor antagonists, and sildenafil has
yet to be determined.
K.M.Kerr,MD,P.F.Fedullo,MD,and W.R.Auger Chronic thromboembolic
Pulmonary Hypertension:When to Suspect It,When to Refer fro
Surgery,Advances in Pulmonary Hypertension,J.Pulmonary Hypertension,V2,No1,pp4-10
Atrial septostomy is a palliative procedure used mainly in
parts of the world where more expensive therapies such as epoprostenol
or lung transplantation are unavailable. By creating a right-to-left
shunt, intentional perforation of the atrial septum unloads
the right ventricle and may increase the patient's functional
capacity. If the patient survives the procedure--associated
mortality is 25%--arterial oxygenation declines, but the increase
in cardiac output usually more than compensates.
Lung transplantation to treat severe pulmonary hypertension
saw a rapid increase in use during the early 1990s but has since
plateaued. Heart-lung transplantation was the primary surgery
during the early 1980s; after advances in techniques to promote
healing of the bronchial anastomosis, single or double lung
transplantations are now preferred at most centers worldwide.
Initial functional improvements following transplantation are
quite impressive, but many patients are not candidates, including
those over age 60, those with significant comorbidity, and those
with deficient psychological or financial resources. The greatest
limitation, however, has been the shortage of donor organs,
with waiting lists exceeding two years at many centers. Since
an anticipated survival of less than 18 months is a selection
criterion at some centers, many patients die while awaiting
transplantation. Considering that bronchiolitis obliterans with
severe airway obstruction eventually develops in a third of
transplant patients and that survival after transplantation
averages only three to five years, up to 70% of patients at
some centers remove themselves from transplantation lists after
responding favorably to intravenous epoprostenol.
Prognosis
According to the National Institutes of Health registry that
followed patients during the 1980s, before the advent of effective
therapies, the natural history of severe pulmonary hypertension
is one of relentless progression to death: One-, three-, and
five-year survival after diagnosis averaged 68%, 48%, and 34%,
respectively; median survival was 2.8 years. Factors that predict
an adverse outcome are poor functional status, high PA pressure
(>80 mm Hg systolic), elevated right atrial pressure (>20
mm Hg), and low cardiac index (<2.0 L/min/m2).
What is the prognosis for patients who respond to vasodilator
therapy? In the short-term, they can expect improved outcomes.
Long-term randomized controlled trials have not been done. Nevertheless,
given the prospect of safer and potentially more effective pharmacotherapies--not
only for primary but for secondary disease as well--clinicians
and patients are justified in harboring even greater optimism
for the future.
PATRICIA M. RUSSO-MAGNO,NICHOLAS S. HILL,Brown University
http://www.hosppract.com/issues/2001/03/russo.htm
PULMONARY HYPERTENSION AND RIGHT HEART FAILURE
Pulmonary hypertension is defined as a mean pulmonary artery
pressure greater than 22 mm Hg. Pulmonary hypertension can occur
from several physiologic causes and disease processes (Table
82); the hypertension may be transient, as in reversible conditions
such as an asthma attack, or chronic, as in emphysema. In some
patients, two or more causes may contribute to pulmonary hypertension
(e.g., left ventricular heart failure and pulmonary emboli).
| Table 82. Causes of pulmonary hypertension |
|
| Disease or condition |
Underlying mechanisms |
| Lung diseases, including all forms of restrictive and
obstructive lung conditions |
Hypoxemia; loss of pulmonary blood vessels; acidosis Hypoxemia;
loss of pulmonary blood vessels; acidosis |
| Lung diseases, including all forms of restrictive and
obstructive lung conditions |
Increased pulmonary capillary hydrostatic pressure |
| Pulmonary thromboembolic disease |
Pulmonary artery narrowing; loss of pulmonary blood vessels
|
| Pulmonary arteritis |
Pulmonary artery narrowing; loss of pulmonary blood vessels
|
| High altitude |
Hypoxemia |
| Hypoventilation |
Hypoxemia; acidosis |
| Chest wall deformity |
Hypoxemia acidosis; pulmonary artery narrowing |
| Idiopathic |
Loss of pulmonary blood vessels; pulmonary artery narrowing
|
| |
|
Table 82:Causes of pulmonary hypertension
Right heart failure is a decompensated state of the right ventricle
and can result from sustained or severe pulmonary hypertension
of any origin. When the right ventricle is unable to pump its
full cardiac output against the elevated pulmonary pressure,
systemic venous pressure increases and fluid "backs up"
in the systemic veins. Untreated, the patient will manifest
leg edema, ascites, liver engorgement, and weight gain. In the
absence of left ventricular failure, there is no excess fluid
in the alveoli, and the lungs will remain clear on chest xray.
A chest xray from a patient with rightside heart failure
is shown in Fig. 81; note the cardiomegaly and the absence
of pulmonary infiltrates. Treatment of right heart failure attempts
to relieve the pulmonary hypertension and uses low sodium intake
and diuretic therapy to help mobilize excess body fluid.

Fig. 81. Chest xray of a patient with pulmonary hypertension
and rightsided heart failure. Note the enlarged heart (caused
by an enlarged right ventricle), the enlarged pulmonary arteries,
and the absence of lung infiltrates .
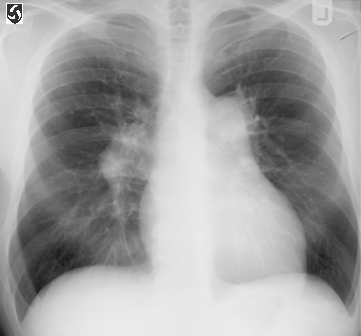
CXR.fig1:PA film of chest in primary pulmonary hypertension
showing right heart enlargement and enlargement of the main
pulmonary artery and its right and left branches.
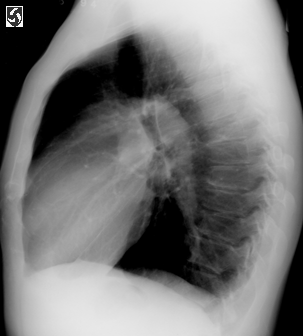
Left lateral CXR.fig2,same patient as above,illustrating the
enlarged pulmonary artery.
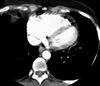
Pulmonary hypertension-fig2:CT of chest showing right heart
enlargement and straightening of the interventricular septum
due to the volume and pressure overload in primary pulmonary
hypertension.
CAUSES OF PULMONARY HYPERTENSION
Lung disease, a common cause of pulmonary hypertension, usually
operates through one of the mechanisms listed in Table 82.
Hypoxemia, a frequent manifestation of lung disease, is one
of the most common physiologic mechanisms causing pulmonary
hypertension. Fig. 82 demonstrates the effect of hypoxemia
on mean pulmonary artery pressure, as well as demonstrating
the interrelationship with acidosis. At normal pH, the arterial
percent saturation of hemoglobin with oxygen SaO2) must decline
to approximately 75% to achieve a doubling of mean pulmonary
artery pressure. When pH is 7.3, the same doubling of pulmonary
artery pressure occurs when the SaO2 is approximately 82%.

Fig. 82. Effect of hypoxemia (reduced SaO2) and acidosis
on mean pulmonary artery pressure. Percentages refer to SaO2.
See text for discussion. (From Mathay, R.A., and Berger, H.J.
: Cardiovascular performances in chronic obstructive pulmonary
diseases, Med. Clin. North Am. 65(3):489524, 1981; reprinted
with permission from W.B. Saunders Co. Reproduced from J. Clin.
Invest. 43:11461162, 1964, by copyright permission of the
American Society for Clinical Investigation.)
Both hypoxemia and acidosis cause pulmonary hypertension by
constricting the small, muscular pulmonary arteries (those less
than 0.2 mm in diameter). The exact mechanism for the vasoconstriction
is unknown. The vasoconstriction may be caused by hypoxia
or acidosismediated release of vasoactive substances or
by a direct effect on pulmonary artery smooth muscle.
|
SIGNS OF COR PULMONALE
Physical examination increased intensity of second
(pulmonic) heart sound; right ventricular heave when palpating
anterior chest wall
Chest xray film enlargement of pulmonary arteries
and right ventricular dilation
Electrocardiogram evidence of rightsided heart
strain, such as tall R wave in precordial leads or tall,
peaked P wave in lead II ( Fig. 83)
|
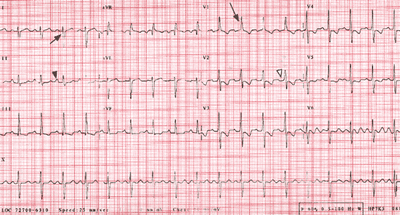
Fig.8-3(ECGPPHTN-fig8)Web: http://www.who.int/ncd/cvd/pph.htm

Fig. 83. ECG readings. A, An example of Ppulmonale
(large peaked P waves in lead 11 [black arrowhead]), which represents
right atrial dilation that results from increased pulmonary
artery and right ventricular pressures. B, A normal ECG.
Hypoxemia is a clinically important cause of pulmonary hypertension
because it is potentially reversible. Continuous oxygen therapy
does reduce mortality from hypoxemic chronic obstructive pulmonary
disease
.
Another cause of pulmonary hypertension is the loss of pulmonary
vasculature. Patients with severe emphysema can actually have
near normal PaO2 yet manifest severe pulmonary hypertension
because the destruction of lung tissue in emphysema may remove
both alveoli and pulmonary capillaries. The remaining lung has
mostly highventilation per fusion ratios that lead to increased
dead space but not to significant hypoxemia . However, since
there is a less vascular bed through which the right ventricle
can pump its cardiac output, the pulmonary artery pressure is
increased.
Cor pulmonale refers to any right ventricular manifestation
of pulmonary hypertension caused by lung disease. Cor pulmonale
usually manifests as one or more signs of rightsided heart
strain-the effects of pulmonary hypertension on the right
ventricle or right atrium . Cor pulmonale is not synonymous
with right heart failure. Of course, the basic cause of cor
pulmonale, pulmonary hypertension, may also lead to rightsided
heart failure.
Perhaps the most common cause of pulmonary hypertension is
left heart failure. (The most common causes of left heart failure
are arteriosclerosis and systemic hypertension.) In left heart
failure fluid backs up in the left atrium and in the pulmonary
circulation, resulting in increased pulmonary artery pressures.
Treatment is usually with digoxin and diuretics and is directed
at the left ventricle. Unless the patient is hypoxemic, supplemental
oxygen can be expected to have little benefit.
Mitral valve disease can cause profound heart failure and pulmonary
hypertension by interfering with the flow of blood from the
left atrium to the left ventricle; this interference can occur
either through mitral stenosis (narrowing of the mitral orifice)
or mitral regurgitation (ejection of blood back into the atrium
during systole). Both conditions are easily diagnosed using
noninvasive cardiac methods and are potentially correctable
with mitral valve surgery. Years ago rheumatic fever was the
principal cause of severe mitral valve disease. Rheumatic heart
disease is now relatively uncommon in the United States, and
as a consequence, the prevalence of severe mitral valve disease
has decreased over the years. Nonetheless, mitral valve disease
should always be considered when pulmonary hypertension is present
without an obvious cause.
Pulmonary emboli are clots that usually arise in the deep veins
of the thigh and pelvis, break off, and travel to lodge in one
or more of the pulmonary arteries. If not fatal to the patient,
these clots will usually dissolve with time; on occasion they
organize and thrombose in situ. Both acute pulmonary emboli
and pulmonary thrombi (emboli that organize and do not dissolve)
are potential causes of pulmonary hypertension. Pulmonary embolism
is a relatively common clinical condition and should always
be considered as a cause of otherwise unexplained pulmonary
hypertension.
Other, rarer causes of pulmonary hypertension are congenital
heart disease, pulmonary arteritis (inflammation of the pulmonary
arteries), and chest wall deformity. Within each category listed
in Table 82 are many different disease entities, far too
numerous to mention.
Pulmonary hypertension may also be of completely unknown origin
(idiopathic). Idiopathic pulmonary hypertension has a predilection
for young and middleaged women and usually presents with
the insidious onset of dyspnea. Diagnosis is made by catheterization
of the right side of the heart, measurement of pulmonary artery
pressures, and by ruling out all other possible causes (e.g.,
heart and lung disease). There is no effective treatment for
this disorder, although several vasodilators have been tried
on an experimental basis. Idiopathic pulmonary hypertension
is usually fatal within 5 years from the time of diagnosis.
ASSESSMENT OF HEMODYNAMIC STATUS
Hemodynamic status refers to the status of the pressure and
the flow within the pulmonary and systemic circulation. Patients
manifesting shock. heart failure, pulmonary hypertension, fluid
overload, and many other problems have altered hemodynamic status.
In clinical practice, there are two levels of hemodynamic assessment.
The first level is noninvasive, meaning without cardiac catheterization
or arterial pressure monitoring. Noninvasive hemodynamic assessment
includes the history, physical examination, chest xray
studies. pulmonary function tests, arterial blood gas measurement,
observation of the patient's response to treatment and, occasionally,
noninvasive heart studies such as the echocardiogram. In the
vast majority of respiratory patients, hemodynamic status can
be assessed noninvasively.
The second level of hemodynamic assessment is invasive and
requires cardiac catheterization and arterial pressure monitoring.
Until the early 1970's, catheterization was only possible in
a special laboratory, and studies were usually limited to noncritically
ill patients with valvular or coronary disease. The advent of
the SwanGanz catheter, first introduced in 1970, made bedside
catheterization feasible and revolutionized hemodynamic evaluation.
In practice, most patients requiring bedside catheterization
also have a small cannula inserted in a peripheral artery (usually
radial) for continuous blood pressure monitoring. In addition,
cardiac rate and rhythm are continuously monitored in all catheterized
patients.
http://www.emedicine.com/MED/topic1962.htm
Pulmonary Hypertension, Primary by Ronald J Oudiz, MD
Last Updated: October 23, 2002
Medical Care:
Anticoagulation
Several studies, using both univariate and multivariate analyses,
show that survival is increased when the patient is treated
with anticoagulant therapy, regardless of histopathologic subtype.
Use warfarin to maintain an International Normalized Ratio
of 1.5- to 2-times the control value, provided no contraindication
to anticoagulation is present.
Other oral agents
Use digoxin therapy to improve right ventricular function in
patients with right ventricular failure. However, no randomized
controlled clinical study has been performed to validate this
strategy for patients with PPH.
Use diuretics to manage peripheral edema.
Use oxygen supplementation in those patients with resting or
exercise-induced hypoxemia. Use caution if patients have a left-to-right
shunt via a PFO (see Imaging Studies) because supplemental oxygen
in these instances may provide little or no benefit.
Conventional oral vasodilator therapy
CCBs are the most widely used class of drugs for PPH. These
drugs are thought to act on the vascular smooth muscle to dilate
the pulmonary resistance vessels and lower the pulmonary artery
pressure. Several studies report clinical and hemodynamic benefits
from the use of long-term calcium channel blockade. The use
of these drugs produces a reduction in pulmonary vascular resistance
(PVR) by increasing the cardiac output and decreasing pulmonary
artery pressure. It also improves the quality of life and survival
rate.
Only use CCBs on patients without overt evidence of right heart
failure. A cardiac index of less than 2 L/min/m2 or elevated
right atrial pressure above 15 mm Hg is evidence that CCBs may
worsen right ventricular failure and, thus, are of no benefit.
This is potentially harmful to patients with PPH.
In general, high doses of CCBs are used in PPH; however, only
patients with an acute vasodilator response to an intravenous
or inhaled pulmonary vasodilator challenge (eg, with adenosine,
epoprostenol [EPO], nitric oxide [NO]) derive any long-term
benefit from CCBs (this corresponds to approximately 25% of
patients with PPH).
Similarly, patients without an acute vasodilator response to
a vasodilator challenge have a worse prognosis on long-term
oral vasodilator therapy compared to those with an initial response.
Importantly, realize that the absence of an acute response
to intravenous or inhaled vasodilators does not preclude the
use of intravenous vasodilator therapy. In fact, continuous
intravenous vasodilator therapy is strongly suggested for these
patients because CCBs are contraindicated.
This illustrates the importance of performing vasoreactivity
testing in patients with PPH. Intravenous EPO or adenosine or
inhaled NO are most commonly used for acute vasodilator testing.
Oxygen, nitroprusside, and hydralazine should not be used as
pulmonary vasodilator testing agents.
Only up to 25% of patients with PPH demonstrate significant
pulmonary vasoreactivity. If patients demonstrate vasoreactivity
and are candidates for high-dose CCB therapy, administer a CCB
challenge to stable patients to determine the vasodilator response.
Perform this in the critical care unit with a balloon flotation
catheter in the pulmonary artery. Administer oral nifedipine
every hour (diltiazem can be used if resting tachycardia is
present) until a 20% decrease in pulmonary artery pressure and
PVR is observed or systemic hypotension or other adverse effects
preclude further drug administration. Calculate the daily dosage
requirement at half the total initial effective dose, and administer
this every 6-8 hours. Typical doses of nifedipine and diltiazem
can reach 240 mg/d and 900 mg/d, respectively. Use caution when
withdrawing CCBs because rebound pulmonary hypertension has
been reported with the cessation of vasodilator therapy.
Three approved pulmonary vasodilator therapies currently available
for PPH are as follows:
Epoprostenol (Flolan)
Treprostinil (Remodulin)
Bosentan (Tracleer)
EPO and treprostinil are given parenterally (see Medication),
and bosentan is given orally.
Future therapies
Clinical trials are underway to determine the safety and efficacy
of several new drugs that include inhaled therapy (ie, prostacyclins,
NO) and orally active drugs.
Efforts are currently focused on prostacyclin analogues, endothelin
antagonists, phosphodiesterase (PDE-5) inhibitors, and thromboxane
inhibitors.
Surgical Care:
A single-lung or double-lung transplant is indicated for patients
who do not respond to medical therapy. Simultaneous cardiac
transplantation may not be necessary even with severe right
ventricular dysfunction; however, this is institutionally dependent.
Diet:
No specific diet is recommended; however, a low-sodium diet
is recommended for those with significant volume overload due
to right ventricular failure.
L-arginine supplementation (a precursor to NO) may be helpful;
however, more studies are needed to confirm its role in the
management of PPH.
Activity:
Few data are available on cardiopulmonary rehabilitation. The
generally accepted recommendation is that patients with heart
failure perform mild symptom-limited aerobic activity and avoid
complete bed rest. Isometric exercises (weight-lifting) are
contraindicated.
Current vasodilator therapy allows for maintenance of a low
PVR in healthy subjects. Three such substances have received
much attention. These are prostacyclin (ie, PGI2, EPO), treprostinil
(Remodulin, a PGI2 analogue), and NO. These molecules are produced
primarily in the vascular endothelium and cause pulmonary vasodilation.
Alterations in the intima of the pulmonary vessels may contribute
to endothelial dysfunction, thereby affecting the release of
PGI2 and NO.
More recently, the ERA bosentan has been approved for initial
PPH therapy in patients with NYHA class III and IV symptoms.
This endothelially active agent improves exercise capacity and
shows promise in halting or reversing pulmonary vascular insult.
Drug Category: Parenteral vasodilators -- Failure to respond
to CCBs or inability to tolerate CCBs with NYHA types III and
IV right heart failure.
Drug Name
|
Epoprostenol (Flolan) -- An analogue of PGI2 that was
approved by the FDA in 1995 for use in patients with PPH.
Has potent vasodilatory properties, an immediate onset of
action, and a half-life of approximately 5 min. In addition
to its vasodilator properties, also contributes to inhibition
of platelet aggregation and plays a role in inhibition of
smooth muscle proliferation. Latter effect may have implications
for beneficial remodeling of pulmonary vascular bed. EPO
is only FDA-approved medication for treatment of PPH. |
| Adult Dose |
Continuous IV infusion via permanent indwelling central
venous catheter using a small, battery-powered infusion
pump worn at the hip or carried in a backpack
Beginning dose: 2-4 ng/kg/min; depending on initial response;
initiate under close observation in the ICU with right heart
flotation catheter in place
Subsequent dose: titrate based on follow-up outpatient evaluation;
common for doses to exceed 40 ng/kg/min after 1 y of therapy
in some patients; currently, no upper limit has been defined
for EPO dosing |
| Pediatric Dose |
Administer as in adults |
| Contraindications |
Documented hypersensitivity; hyaline membrane disease,
dominant left-to-right shunt, respiratory distress syndrome |
| Interactions |
Coadministration with anticoagulants may increase bleeding
risk due to shared effects on platelet aggregation |
| Pregnancy |
B - Usually safe but benefits must outweigh the risks.
|
| Precautions |
Coadminister with anticoagulants whenever possible to
reduce risk of thromboembolism; sudden discontinuation or
reduction in therapy may result in rebound pulmonary hypertension |
| Drug Name |
Treprostinil (Remodulin) -- Used to treat PAH. Structurally
very similar to EPO but stable at room temperature and has
much longer half-life; therefore, can be given as an SC
continuous infusion via a much smaller pump. Elicits direct
vasodilation of pulmonary and systemic arterial vessels
and inhibits platelet aggregation. Vasodilation reduces
right and left ventricular afterload and increases cardiac
output and stroke volume. |
| Adult Dose |
1.25 ng/kg/min SC via continuous infusion initially; may
increase by 1.25 ng/kg/min each wk for 4 wk, then may increase
by 2.5 ng/kg/min each wk; not to exceed 40 ng/kg/min
Note: If initial dose not tolerated, decrease to 0.625 ng/kg/min,
then slowly titrate upward; must slowly taper if discontinued
(potential for severe rebound pulmonary hypertension and
death |
| Pediatric Dose |
Not established |
| Contraindications |
Documented hypersensitivity |
| Interactions |
Additive hypotensive effect with antihypertensive agents
or diuretics; may increase risk of bleeding with other antiplatelet
drugs (eg, aspirin) or anticoagulants (eg, warfarin, heparin) |
| Precautions |
B - Usually safe but benefits must outweigh the risks.
|
| |
|
| |
|
Drug Category: Oral pulmonary hypertension agents -- ERAs and
are alternative therapy to parenteral prostacyclin agents. Given
PO. Competitively bind to endothelin-1 (ET-1) receptors endothelin-A
and endothelin-B, causing reduction in PAP, PVR, and mean RAP.
Indicated for treatment of PAH in patients with WHO class III
or IV symptoms to improve exercise ability and decrease rate
of clinical deterioration.
Drug Name
|
Bosentan (Tracleer) -- First oral PPH therapy to gain
approval in United States. A mixed endothelin-A and endothelin-B
receptor antagonist indicated for the treatment of PAH,
including PPH. In clinical trials, improved exercise capacity,
decreased rate of clinical deterioration, improved functional
class, and improved hemodynamics.
Improves pulmonary arterial hemodynamics by competitively
binding to ET-1 receptors endothelin-A and endothelin-B
in pulmonary vascular endothelium and pulmonary vascular
smooth muscle. This leads to a significant increase in CI
associated with a significant reduction in PAP, PVR, and
mean RAP. These changes result in an improvement in exercise
capacity (as measured by the 6-min walk test) and improved
PPH symptoms.
Because drug has teratogenic potential and because of need
for careful scrutiny in choosing appropriate candidates
for ERA therapy, Tracleer can be prescribed only through
the Tracleer Access Program. Call 1-866-228-3546. |
| Adult Dose |
Starting dose: 62.5 mg PO bid for 4 wk, followed by 125
mg PO bid indefinitely |
| Pediatric Dose |
Not established; 62.5 mg PO bid recommended if <40
kg or >12 y; not to exceed 125 mg/d |
| Contraindications |
Documented hypersensitivity; coadministration with cyclosporine
A or glyburide |
| Interactions |
Toxicity may increase when administered concomitantly
with inhibitors of isoenzymes CYP450 2C9 and CYP450 3A4
(eg, ketoconazole, erythromycin, fluoxetine, sertraline,
amiodarone, cyclosporine A); induces isoenzymes CYP450 2C9
and CYP450 3A4, causing decrease in plasma concentrations
of drugs metabolized by these enzymes (including glyburide
and other hypoglycemics, cyclosporine A, hormonal contraceptives,
simvastatin, and possibly other statins); hepatotoxicity
increases with concomitant administration of glyburide
Regarding cyclosporine A, during first day of concomitant
administration, trough concentrations of bosentan were increased
approximately 30-fold; steady-state bosentan plasma concentrations
were 3- to 4-fold higher than in the absence of cyclosporine
A
Regarding glyburide, an increased risk of elevated liver
aminotransferase levels was observed in patients receiving
concomitant therapy with glyburide |
| Pregnancy |
X - Contraindicated in pregnancy |
| Precautions |
May cause a dose-related decrease in hemoglobin and hematocrit;
hemoglobin levels should be monitored after 1 and 3 mo of
treatment and then every 3 mo; overall mean decrease in
hemoglobin concentration was 0.9 g/dL (change to end of
treatment); most of this decrease of hemoglobin concentration
was detected during first few weeks of treatment, and hemoglobin
levels stabilized by 4-12 wk of treatment
In placebo-controlled studies of all uses of bosentan, marked
decreases in hemoglobin (>15% decrease from baseline,
resulting in values <11 g/dL) were observed in 6% of
bosentan-treated patients and 3% of placebo-treated patients;
in patients with PAH treated with doses of 125 and 250 mg
bid, marked decreases in hemoglobin occurred in 3% of bosentan-treated
compared to 1% in placebo-treated patients
A decrease in hemoglobin concentration by at least 1 g/dL
was observed in 57% of bosentan-treated patients, as compared
to 29% of placebo-treated patients; in 80% of those patients
whose hemoglobin decreased by at least 1 g/dL, the decrease
occurred during the first 6 wk of bosentan treatment
During the course of treatment, hemoglobin concentration
remained within normal limits in 68% of bosentan-treated
patients compared to 76% of placebo patients (explanation
for change in hemoglobin is not known, but hemorrhage or
hemolysis do not appear to be the cause)
Check hemoglobin concentrations after 1 and 3 mo and every
3 mo thereafter; if a marked decrease in hemoglobin concentration
occurs, further evaluation should be undertaken to determine
cause and need for specific treatment
Causes at least 3-fold elevation of liver aminotransferase
levels (ie, ALT, AST) in up to 11% of patients; may elevate
bilirubin (serum aminotransferase levels must be measured
prior to initiation of treatment and then monthly); caution
in patients with mildly impaired liver function (avoid in
patients with moderate or severe liver impairment)
Not recommended while breastfeeding; exclude pregnancy before
initiating treatment and prevent thereafter by use of reliable
contraception
Headache and nasopharyngitis may occur
FOLLOW-UP
|
Further Inpatient Care:
Patients on EPO therapy must have a central venous catheter
placed surgically and receive their initial EPO dose in an inpatient
setting. This allows for monitoring of acute adverse effects
and provides the opportunity for the patient and support personnel
to master the EPO preparation and administration technique before
discharge.
Further Outpatient Care:
Currently, no precise dosage adjustment algorithm is available
for patients with PPH who are on vasodilator therapy.
Monitor the patient with frequent physical examinations, and
focus the history on heart failure symptoms and adverse effects
of medications.
Echocardiography has been used in several studies to serially
monitor changes in the right ventricular–right atrial pressure
gradient and the right and left ventricular chamber size.
Findings from other noninvasive modalities (eg, electron-beam
CT measurements of cardiac chamber sizes) correlate with hemodynamic
improvements in pulmonary physiology.
More recently, cardiopulmonary exercise testing, serial invasive
hemodynamic testing, and 6-minute walk testing have been used
to monitor the disease status of patients with PPH.
Complications:
Advanced right heart failure with hepatic congestion
Pedal edema
Pleural effusions
Ascites
Worsening dyspnea on exertion
Prognosis:
The mortality rate for untreated PPH is approximately 50% at
3 years. With EPO therapy, this has increased to higher than
65% at 5 years. Patients whose disease progresses either undergo
transplantation or die of progressive right heart failure.
Patient Education:
Patient and physician education about this rare fatal disease
is paramount.
In addition, instruct the patient on how to perform EPO therapy
(ie, how to mix and administer their IV medication on a daily
basis).