MANAGEMENT
OF MYOCARDIAL INFARCTION
(HEART ATTACK) |
Objects of early treatment
1). Provide cardiac
resuscitation if necessary.
2). Immediate
hospitalization.
3). Treat life
threatening arrhythmias.
4). Alleviate
pain and suffering.
5). Preserve
as much myocardium as possible by the following:
Cardioprotection by Increasing Coronary Flow:Thrombolysis
(Dissolution of Blood Clot or Thrombus Blocking Lumen of Coronary
Artery)
Thrombolysis is based on the following
facts:
A). The evolution
of myocardial necrosis (death) following coronary occlusion
is time-dependent over 4-6 hours.
B). The restoration
of coronary blood flow within this 4-6 hour period salvages
myocardium, but in a time related fashion.
C). The proximate
cause of myocardial infarction is generally an occlusive thrombus
within the coronary artery.
Clinical trials have shown that Early Reperfusion
Induced by a Thrombolytic Agent --- Recombinant
Tissue Plasminogen Activator, Streptokinase, Urokinase, or Anisoylated
Plasminogen Streptokinase Activator Complex --- is associated
with Limitation of Infarct Size, Preservation of Ventricular
Function, and Improved Survival.
Sustained Patency and the Impact of
Reocclusion
The incidence of coronary reocclusion from
rethrombosis is about 20 per cent, and the incidence of reinfarction
is about 10 per cent, occurring mainly in the first 24 hours.
Contraindications to Thrombolytic Therapy
1). Cerebral
vascular accident (stroke) within the last 3 months
2). Recent major
surgery
3). Pregnancy,
aortic dissection, recent head trauma, brain tumor
Risks of Thrombolytic Therapy: Possible
Intracranial Bleeding (Stroke)
Conjunctive and Adjunctive Therapy
1). Heparin (an
anticlotting drug) is given intravenously to prevent rethrombosis
and reocclusion: 5000 units as a bolus, followed by an infusion
of 1000 to1200 units/h to keep the PTT (a measure of the clotting
time of blood) at 1.5 to2.0 times normal for the first 24-48
hours.
2a). Aspirin is
more effective than heparin after 24 to 48h, because the plasma
thrombin levels have returned to normal by this time. Aspirin
reduces the incidence of reocclusion and reinfarction. It should
be given as soon as possible and continued indefinitely.
2b). The antiplatelet
agent clopidogrel has been found to lower the number of nonfatal
myocardial infarction or stroke in patients with acute coronary
syndromes without ST-segment elevation to 9.3% compared to placebo
and aspirin with 11.4% over a 3 to 12 month period. Also, the
above or refractory ischemia occurred in 16.5% in the clopidogrel
group compared to 18.8 % in the placebo group. But there were
significantly more patients with major bleeding in the clopidogrel
group (3.7%) than in the placebo group (2.7%), but there were
not significantly more patients with life-threatening bleeding
(2.1% versus 1.8%) or hemorrhagic stroke.
Reference:Clopidogrel in Unstable Angina to
Prevent Recurrent Events trial Investigators,Effects of Clopidogrel
in Addition to Aspirin in Patients with Acute Coronary Syndromes
Without ST-Segment Elevation,N Engl J Med,Vol.345,No.7,August
16,2001 ,Pp.494-502.
3). Beta-Adrenergic
Blockers ( medications like metoprolol, atenolol) effects :
a) Reduces ventricular
ectopy, atrial fibrillation, and nonfatal cardiac arrest
b) Reduces frequency
of progression of threatened infarction to completed infarction
c) Reduces recurrent
ischemia and infarction during first 6 weeks after initial event
4). Magnesium
has the following effects:
a) reduces all
causes of death and coronary care mortality rates;
b) reduces congestive
heart failure in the coronary care unit;
c) preserves myocardium;
d) may reduce
ventricular arrhythmias.
Angioplasty and Surgical Revascularization as Primary or Adjunctive
Therapy to ThrombolysisRisk of Thrombolytic Therapy: Hemorrhagic
strokengioAdjunctive Theray to Thrombolyses in which angioplasty
may be per
1). Direct angioplasty
as the primary reperfusion technique.
2). Immediate
angioplasty after thrombolytic agent.
3). Rescue angioplasty
if thrombolysis fails.
4). Delayed angioplasty
routinely after thrombolysis.
5). Elective
angioplasty if ischemia occurs after reperfusion.
Use of Glycoprotein IIb/IIIa Inhibitors
A recent study has shown that the use of the
platelet glycoprotein IIb//IIIa inhibitor tirofiban in patients
with unstable coronary syndromes (unstable angina and myocardial
infarction without ST elevation) and early invasive strategy
including percutaneous coronary revascularization and intracoronary
stents reduce the risk of death, myocardial infarction or recurrent
angina. The benefit of the early invasive strategy was greater
in patients with troponin T levels (markers of myocardial injury)
of more than 0.01ng per milliliter than in patients with levels
of 0.01ng per milliliter or less.
The benefits of early invasive strategy, similarly,
were largely confined to patients with ST-segment depression
on the admission electrocardiogram. The advent of "point
of care" measurements of troponin levels in the emergency
room, together with the routine electrocardiogram, has made
the goal of rapid identification of high risk patients most
likely to benefit from an early invasive approach readily attainable.
It is thus unnecessary to recommend the use of such an approach
for all patients with acute coronary syndromes, especially those
without elevated levels of cardiac enzymes or troponins or ST-segment
depression.
Reference:Cannon,C.P.,M.D. and others,Comparison
of early invasive and conservative strategies in patients wit
unstable coronary syndromes treated with the glycoprotein IIb/IIIa
inhibitor tirofiban,N.Engl.J.Med.,Vol.344,No.25.June 21,2001,PP.1879-1887.
Reference:Boden,W.E. andMackay,R.G.,Optimal
treatment of acute coronary syndromes-An evolving strategy,N.Engl.J.Med.,Vol.344,No.25.June
21,2001,PP.1939-1942.
Another study showed that tirofiban offerred
less protection from major ischemic events with percutaneous
coronary revascularization than did abciximab, another platelet
inhibitor.
Reference:Topol,E.J. and others,Comparison
of two platelet glycoprotein IIa/IIIb inhibitors,tirofiban and
abciximab, for the prevention of ischemic events with percutaneous
coronary revascularization,N.Engl.J.Med.,Vol.344,No.25.June
21,2001,PP.1888-1893
There is also a recent report that early administration
of abciximab in patients with acute myocardial infarction improves
coronary patency before stenting, the success rate of stenting
procedure, and the rate of coronary patency at six months, left
ventricular function, and clinical outcomes.
Reference:Montalescot,G. and others,Platelet
glycoprotein IIb/IIIa inhibition with coronary stenting for
acute myocardial infarction,N.Engl.J.Med.,Vol.344,No.25.June
21,2001,PP.1895-903.
Troponin Measurements in Ischemic Heart
Disease
Current nomenclature categorizes patients
with ischemic discomfort into those who present with ST elevation
of the twelve lead electrocardiogram (E CG) versus those who
do not present with ST elevation. Patients presenting with ST-segment
elevation are, of course, easy to recognize using the ECG. The
majority of such patients will develop a Q wave on the ECG and
will ultimately be diagnosed as having sustained a Q-wave myocardial
infarction(MI). Patients who present without ST elevation are
experiencing either unstable angina(UA) or a non-ST- segment
elevation MI (NSTEMI). Most patients who present with NSTEMI
do not develop a Q wave on the ECG. The distinction between
unstable angina and NSTEMI is based on the presence or absence
of a cardiac marker in the blood. When such a cardiac marker
is detected in a patient's blood, the patient is ultimately
diagnosed as having a non-Q wave myocardial infarction.
Biochemical markers of myocyte necrosis are
useful not only for making the diagnosis of an MI, but also
with estimating prognosis. Of interst are the macromolecules
that diffuse out of necrosing myocytes as membrane integrity
is lost. Once outside the myocyte, the macromolecules are cleared
from the interstitium by cardiac lymphatics. Eventually when
the capacity of lymphatics to clear the macromolecules is exceeded,
the markers become detectable in the peripheral circulation.
Both clinical chemical laboratory of assays and bedside assays
are available to measure several biochemical cardiac markers,
notably myoglobin and the MB fraction of creatine kinase( CK-MB)
and cardiac specific troponins T and I (cTnT and cTn I).
Although myoglobin and CK.-MB are familiar
to physicians and provide reasonable sensitivity for detecting
MI, they lack specificity in the setting of skeletal muscle
disease or injury. Hence, there is intense interest in cTnT
and cTn I as markers that are found in high concentration in
the myocardium and are released with a stoichiometric relationship
proportional to the amount of myocardial injury. Monoclonal-antibody-based
assays are available that capitalize on the fact that the amino
acid sequences of the cardiac and skeletal muscle forms of troponins
T and I are sufficiently dissimilar. After much debate and discussion
expert panels have declared cTnT and cTnI to be the preferred
biomarkers for detection of myocardial damage.
Myocardial necrosis (i.e.MI) is said to be
present if the maximal concentration of cTnT or cTnI exceeds
the decision limit (99 percent of the values for a reference
control group) on at least one occasion during the 24 hours
after the index clinical event. It is important to note that
patients may have an episode of microinfarction where cTnT or
cTnI measurements exceed the decision limits and yet CK-MB even
by mass assay remains in the normal range. It is estimated that
about 1/3 of patients presenting without ST elevation who would
previously have been diagnosed as experiencing unstable angina
on the basis of normal CK-M B levels are now diagnosed as experiencing
NSTEMI because of detectable troponin levels.
In a recent study the results of several
trials reported that the relative risk of troponin positive
patients (compared with troponin negative patients)was 3.9 for
mortality and 3.8 for death or nonfatal recurrent MI. The adverse
prognostic significance of a positive troponin test has been
demonstrated by multiple chemical investigators across multiple
trials, involving multiple patient groups from different countries,
indicating that the observation is a robust one.
Part of the answer to why the patients with
a positive troponin test have a worse prognosis may simply be
that the positive troponin is indicative of myocardial necrosis.
Because it is well established that left ventricular(LV) function
is a pivotal determinant of prognosis, it is possible that loss
of functioning myocardium is associated with a worse outcome.
Several investigators have reported dichotomous analysis showing
that the patients who present with UA/ NSTEMI and a positive
troponin test are significantly more likely to have high-risk
angographic anatomy of the culprit lesion. Troponin-positive
patients are more likely to have visible thrombus in the culprit
lesion,complex lesions, and worse thrombolysis in myocardial
infarction flow grade. It has been argued that these anatomical
findings put troponin-positive patients at risk for downstream
embolization of atherothrombotic debris, occluding the microvasculature
and causing foci of microinfarction-hence the release of troponin.
This line of reasoning leads to the oft-repeated
recommendation by several investigators that patients with positive
troponin test should be treated aggressively with antithrombotic
therapy with particular emphasis on the intravenous IIb/IIIa
inhibitors. It also been argued that troponin-positive patients
should be selected for referral for early invasive strategy.
Clearly ,not all troponin positive patients are at the same
level of risk. Note that there is a highly significant gradient
of increasing risk of mortality with increasing troponin levels.
In a recent study patients with increasing cTnT levels tended
to have progressively greater delays from the onset of symptoms
to blood sampling, were significantly more likely to present
a ST depression or abnormal Q-waves on their ECG. And were progressively
more likely to have depressed LV function on echocardiography
(ejection fraction less than 45 percent). In the subset patients
who were randomized to an early invasive strategy, angiography
did not show any correlation between the severity of underlying
coronary artery disease in the cTnT level but did show a progressive
increase in the odds of visible thrombus and a progressive decrease
in the odds of TIMI flow grade 3 with increasing cTnT levels.
Complementing the angiographic finding from
the early invasive strategy are the interesting clinical findings
in the early noninvasive cohort. The 12-month mortality was
numerical lowest in the cTnT negative group and was slightly
higher in the cTnT positive patients, but there was no clear
trend toward increasing mortality with increasing troponin levels
when analyze by tertilesof positive cTnT. However, a U-shaped
relationship was observed between troponin measurements and
the rate of MI through one year;MI occurred in 5.5% of cTnT
negative patients (<0.01 nanogram/ml., 17.5% of patients in
the first tertile of the positive cTnT results (0.01 to 0.17nanogram/ml),
16.2% in the second tertile(o.18 to0.63 nanogram/ml), but only
8.4 % in the third tertile (>0.63ng/ml. A similar U-shaped relationship
was found with referral for revascularization: 38.8%,51.9%,46.1%
and 34.3%, respectively, in the same four groups.
In another study patients with either undetectable
or only slightly elevated levels of cTnT or cTnI showed no benefit
of tirofiban with respect to prevention of death or nonfatal
MI by 30 days. Those with intermediate elevations of either
cTnT or cTnI had the maximal benefit, whereas those with higher
biomarker levels showed progressive less benefit compared with
the intermediately elevated group.
Those patients with negative tests (below
the decision limit) or just barely elevated quantitative results
are at low risk.They are unlikely to benefit from IV glycoprotein
IIb/IIIa inhibitors. In general, they are managed equally well
with either an early conservative or early invasive strategy
(depending on patient and physician preferences). Multiple clinical
factors other than the results of biomarkers should be included
in the decision about referral for revascularization. Those
patients with an intermediately elevated troponin levels have
not yet lost substantial amounts of myocardium and are excellent
candidates for effective antithrombotic therapy (e.g.,enoxaparin
in place of unfractionated heparin) and prompt referral for
an early invasive approach supported by IV glycoproteins inhibitors
in the catheterization laboratory.
Those patients with the highest level of troponin
have already lost substantial amounts of myocardium.It is not
clear it is not so clear that aggressive antithrombotic therapy
with glycoprotein inhibitors will be helpful in such patients.
The focus should be early diagnostic coronary arteriography.
Some patients may have a completed left circumflex infarction
masquerading as / NSTEMI . Others may have severe multivessel
disease or other r high risk coronary anatomy in association
with depressed LV function (as suggested by high troponin levels);
this group is likely to be best served by referral for coronary
artery bypass surgery.
Because of the uncertainty about the best
course of action for patients with highest level of troponin
upstream use of IV glycoproteinIIb/IIIa inhibitors is not favored.
Instead, diagnostic catheterization is advised to determine
whether revascularization is needed, art in the in and if so,
whether referral for surgery is best without IV glycoproteins
or whether a percutaneous intervention supported by the inhibitors
is
Reference:Antman,E.M., Troponin Measurements
in Ischemic heart Heart Disease:More Than Just a Black and White
Picture,AmericanCollege Cardiology ,Vol.38,N.4 2001,PP.987990
ACC/AHA 2002 Guideline Update
for the Management of Patients With
Unstable Angina and Non-ST-Segment
Elevation Myocardial Infarction-Summary Article
A Report of the American College of Cardiology/
American Heart Association Task Force on Practice Guidelines
(Committee on the Management of Patients With Unstable Angina)
COMMITTEE MEMBERS
EUGENE BRAUNWALD, MD, FACC, FAHA, Chair
ELLIOTT M. ANTMAN, MD, FACC, FAHA
JOHN W. BEASLEY, MD, FAAFP
ROBERT M. CALIFF, MD, FACC
MELVIN D. CHEITLIN, MD, FACC
JUDITH S. HOCHMAN, MD, FACC, FAHA
ROBERT H. JONES, MD, FACC
DEAN KEREIAKES, MD, FACC
JOEL KUPERSMITH, MD, FACC, FAHA
THOMAS N. LEVIN, MD, FACC
CARL J. PEPINE, MD, MACC, FAHA
JOHN W. SCHAEFFER, MD, FACC, FAHA
EARL E. SMITH III, MD, FACEP
DAVID E. STEWARD, MD, FACP
PIERRE THEROUX, MD, FACC, FAHA
TASK FORCE MEMBERS
RAYMOND J. GIBBONS, MD, FACC, FAHA,
Chair
ELLIOTT M. ANTMAN, MD,
FACC, FAHA, Vice Chair
JOSEPH S. ALPERT, MD, FACC, FAHA
DAVID P. FAXON, MD,
FACC, FAHA
VALENTIN FUSTER, MD,
PIID, FACC, FAHA
GABRIEL GREGORATOS, MD,
FACC, FAHA
LOREN F. HIRATZKA, MD, FACC, FAHA
ALICE K. JACOBS, MD,
FACC, FAHA
SIDNEY C. SMITH, JR, MD, FACC, FAHA
INTRODUCTION
The American College of Cardiology (ACC)/American
Heart Association (AHA) guidelines for the management of unstable
angina and non-ST-segment elevation myocardial infarction (UA/NSTEMI)
were published in Septemher 2000 (1). Since then, a number of
clinical trials and observational studies have been published
or presented that, when taken together, alter significantly
the recommendations made in that document. Therefore, the ACC/AHA
The ACC/AHA Task Force on Practice Guidelines makes every effort
to avoid any actual or potential conflicts of interest that
might arise as a result of an outside relationship or personal
interest of a member of the writing panel. Specifically, all
members of the writing panel are asked to provide disclosure
statements of all such relationships that might be perceived
as real or potential conflicts of interest. These statements
are reviewed by the parent task force, reported orally to all
members of the writing panel at the first meeting, and updated
as changes occur.
This document was approved by the American
College of Cardiology Foundation Board of Trustees in September
2002 and by the American Heart Association Science Advisory
and Coordinating Committee in August 2002.
When citing this document, the American College
of Cardiology Foundation and the American Heart Association
would appreciate the following citation format: Braunwald E,
Anturan EM, Beasley JW, Califf RM, Cheitlin MD, IIochman JS,
Jones RI- I, Kereiakes D, Kupersmith J, Levin TN, Pepine CJ,
Schaeffer JW, Smith EE III, Steward DE, Theroux P.ACC/AHA 2002
guideline update for the management of patients with unstable
angina and non-ST-segment elevation myocardial infarction: summary
article: a report of the American College of Cardiology/American
Heart Association Task Force on Practice Guidelines (Committee
on the Management of Patients With Unstable Angina). J Am Coll
Cardiol 2002;40:1366-74.
This document is available on the World Wide
Web sites of the ACC (wow. acc.org) and the AHA (www.americanheartorg).
Single copies of this document arc available for $5 each by
calling 800-253-4636 (US only) or writing the American College
of Cardiology Foundation, Resource Center, 9111 Old Georgetown
Road, Bethesda, MD 20814-1699 (product code 71-0227).This document
and the companion full-text guidelines (product code 71-0240),
are available on the ACC Web site at www.acc.org and the AHA
Web site at www.anieiicauheart.org. To purchase additional reprints
(specify version): up to 999 copies, call 800-611-6083 (US only)
or fax 413-665-2671; 1,000 or more copies, call 214-706-1466,
fax 214-691-6342; or E-mail pubauth©heart.org
Committee on the Management of Patients With
Unstable Angina, with the concurrence of the ACC/AHA Task Force
on Practice Guidelines, revised these guidelines. These revisions
were prepared in December 2001, reviewed and approved, and then
published on the ACC World Wide Web site (www.acc.org) and AHA
World Wide Web site (www.americanheart.org) on March 15, 2002.
The present article describes these revisions and provides further
updates in this rapidly moving field. Minor clarifications in
the wording of three recommendations that now appear differently
from those that were previously published on the ACC and AHA
Web sites are noted in footnotes.
The ACC/AHA classifications I, II, and III are used to summarize
indications as follows:
ClassI:Conditions for which there is evidence and/or general
agreement that a given procedure or treatment is useful and
effective. ClassII:Conditions for which there is conflicting
evidence and/or a divergence of opinion about the usefulness/efficacy
of a procedure or treatment.
IIa: Weight of evidence/opinion is in favor of usefulness/efficacy.
IIb: Usefulness/efficacy is less well established by evidence/opinion.
ClassIII: Conditions for which there is evidence and/or general
agreement that the procedure/treatment is not useful/effective
and in some cases may be harmful.
The weight of the evidence was ranked highest (A) if the data
were derived from multiple randomized clinical trials that involved
large numbers of patients and intermediate (B) if the data were
derived from a limited number of randomized trials that involved
small numbers of patients or from careful analyses of nonrandomized
studies or observational registries. A lower rank (C) was given
when expert consensus was the primary basis for the recommendation.
RISK ASSESSMENT
Clinical Features
Unstable angina and NSTEMI are heterogeneous
disorders in which patients have widely varying risks. Risk
is an important "driver" of management decisions,
and accurate yet simple methods of risk assessment are important
for patient care.
Risk was assessed by multivariable regression
techniques in patients presenting with UA/NSTEMI in several
large clinical trials. Boersma et al. analyzed the relation
between baseline characteristics and the incidence of death
and the composite of death or myocardial (re)infarction at 30
days in patients who entered the PURSUIT (Platelet IIb/IIIa
in Unstable angina: Receptor Suppression Using Integrilin Therapy)
trial (2). The most important baseline features associated with
death were age, heart rate, systolic blood pressure, ST-segment
depression, signs of heart failure, and
elevation of cardiac biomarkers. From this analysis, a simple
risk estimation score was developed.
Antman et al. developed a 7-point risk score,
the "TIMI Risk Score," (age greater than or equal
to 65 years, more than 3 coronary risk factors, prior angiographic
coronary obstruction, ST-segment deviation, more than 2 angina
events within 24 h, use of aspirin [ASA] within 7 days, and
elevated cardiac markers) (3). The score was defined as the
simple sum of these individual prognostic variables. The risk
of developing an adverse outcome-death, (re)infarction, or recurrent
severe ischemia that required revascularizationranged from 5%
with a score of 0 or 1 to 41% with a score of 6 or 7. The score
was derived from data in the TIMI 11B (Thrombolysis In Myocardial
Infarction 11B) trial (4) and then validated in 3 additional
trials-ESSENCE (Efficacy and Safety of Subcutaneous Enoxaparin
in Non-Q-wave Coronary Events study) (5), and PRISM-PLUS (Platelet
Receptor inhibition for Ischemic Syndrome Management in Patients
Limited by Unstable Signs and symptoms) (6) and prospectively
in one TACTICS-TIMI 18 (Treat angina with Aggrastat and determine
Cost of Therapy with an Invasive or Conservative Strategy-Thrombolysis
In Myocardial Infarction) 18 (7). A progressively greater benefit
from newer therapies such as low-molecular-weight heparin (LMWH)
(4,5), platelet glycoprotein (GP) IIb/IIIa receptor antagonists
(6), and an invasive strategy (7) with increasing risk score
have been reported.
Biomarkers
The Joint European Society of Cardiology/American
College of Cardiology Committee for the Redefinition of Myocardial
Infarction (8) emphasized the use of troponins as critical markers
of the presence of myocardial necrosis. Although troponins are
accurate in identifying myocardial necrosis, the latter is not
always secondary to atherosclerotic coronary artery disease.
Therefore, in establishing the diagnosis of NSTEMI, cardiac
troponins should be used in conjunction with appropriate clinical
features and electrocardiographic changes. Myocardial injury
of diverse origins (e.g., myocarditis, trauma, or cardioversion)
may cause necrosis and release of troponins. Although these
may be considered instances of NSTEMI, they should be distinguished
on clinical grounds from the more common form of NSTEMI secondary
to coronary atherosclerosis.
Antiplatelet Therapy
Antiplatelet therapy is a cornerstone
in the management of UA/NSTEMI. Three classes of antiplatelet
drugs (ASA, thienopyridines, and GP Ilb/IIIa antagonists) have
been found useful in the management of these patients and are
the subject of continued intensive investigation and analysis.
Clopidogrel. Given its more rapid onset of action (9,10) and
better safety profile compared with ticlopidine, clopidogrel
is now the preferred thienopyridine. The CURE (Clopidogrel in
Unstable angina to prevent Recurrent ischemic Events) trial
(11) randomized 12,562 patients withUA/STEMI who presented within
24 h to placebo or clopidogrel (loading dose of 300 mg followed
by 75 mg daily) and followed them for 3 to 12 months; all patients
were given aspirin. Cardiovascular death, myocardial infarction
(MI), or stroke occurred in 11.5% of patients assigned to placebo
and 9.3% of those assigned to clopidogrel (relative risk [RR]
0.80; p less than 0.001). Looking at the individual components
of the primary composite and end point, there was a trend in
favor of clopidogrel for cardiovascular death and stroke (5.5%
and 1.4%, respectively, for placebo vs. 5.1% and 1.2% for clopidogrel),
and there was a significant reduction in MI (6.7% vs. 5.2% R.R.
= 0.77, p less than 0.001). However, there was no significant
difference in the incidence of non-Q-wave MI (3.8% vs. 3.5%).
A reduction in recurrent ischemia was noted within the first
few hours after randomization. These salutary results were observed
across all subgroups of patients. There was, however, a significant
excess of major bleeding (2.7% in the placebo group versus 3.7%
in the clopidogrel group; p = 0.003) and of minor bleeding,
as well as a (nonsignificant) trend for an increase in life-threatening
bleeding. The risk of bleeding was increased in patients who
underwent coronary artery bypass grafting (CABG) within the
first 5 days after clopidogrel was discontinued.
The CURE trial was performed in hospitals in which there was
no routine policy of early invasive procedures, and therefore,
revascularization was performed during the initial admission
in only 23% of the patients, a substantially lower percentage
than currently receive this therapy at most US hospitals. Although
the addition of a GP IIb/IIIa antagonist appeared to be well
tolerated in patients who were given ASA, clopidogrel, and heparin
in CURE, fewer than 10% of patients received this combination.
Therefore, additional information on the safety of "quadruple
therapy" (heparin [unfractionated or low molecular weight],
ASA, clopidogrel, and a GP 11b/111a antagonist) should be obtained.
The CURE trial provides strong support for the addition of clopidogrel
to ASA on admission in the management of patients with UA and
NSTEMI. Clopidogrel appears to be especially useful in hospitals
that do not have a routine policy of early invasive procedures
and in patients who are not candidates or who do not wish to
be considered for revascularization. The optimal duration of
therapy with clopidogrel has not been determined. The major
benefits in CURE were observed at 30 days, with small additional
benefits observed over the subsequent treatment period, which
averaged 8 months.
In PCI-CURE, a substudy of CURE, 2,658 patients who underwent
percutaneous coronary intervention (PCI) had been randomly assigned
to double-blind treatment with clopidogrel (n = 1,313) or placebo
(n = 1,345) (12); all patients also received ASA. Patients were
pretreated with placebo or study drug for a median of 10 days
before PCI. After the procedure, most patients received open-label
thienopyridine (clopidogrel or ticlopidine) for approximately
4 weeks, after which the study drug (placebo or
clopidogrel) was again administered for an average of 8 months.
The primary end point, a composite of cardiovascular death,
MI, or urgent target-vessel revascularization within 30 days
of PCI, occurred in 86 patients (6.4%) in the placebo group
compared with 59 (4.5%) in the clopidogrel group (RR 0.70; p
= 0.03). When events that occurred before and after PCI were
considered, there was a 31% reduction in cardiovascular death
or MI with assignment to clopidogrel (p = 0.002). Thus, in patients
with UA and NSTEMI who are given ASA and are undergoing PCI,
a strategy of clopidogrel pretreatment followed by at least
1 month and probably longer-term therapy is beneficial in reducing
major cardiovascular events (12).
There now appears to be an important role for clopidogrel in
patients with UA/NSTEMI, both those who are managed conservatively
and those who undergo PCI, especially stenting. However, it
is not entirely clear how long therapy should be maintained.
Because clopidogrel, when added to ASA, increases the risk of
bleeding during major surgery in patients who are scheduled
for CABG, if possible, clopidogrel should be withheld for at
least 5 days (11) and preferably for 7 days before surgery (13).
In many hospitals in which patients with UA/NSTEMI undergo diagnostic
catheterization within 24 to 36 h of admission, clopidogrel
is not started until it is clear that CABG will not be scheduled
within the next several days. A loading dose of clopidogrel
can be given to a patient on the catheterization table if a
PCI is to be performed immediately. If PCI is not performed,
clopidogrel can be begun after the catheterization.
Glycoprotein IIb/IIIa antagonists in
PCI.
The introduction of platelet GP IIb/IIIa
antagonists represents an important advance in the treatment
of patients with UA/ NSTEMI who are undergoing PCI. These drugs
take advantage of the fact that platelets play an important
role in the development of ischemic complications that may occur
in patients with UA/NSTEMI during coronary revascularization
procedures. The September 2000 guidelines emphasized the value
of GP Ilb/IIIa antagonists in patients with UA/NSTEMI who were
undergoing PCI (1).
Two trials of GP IIb/IIIa inhibitors have been published since
September 2000. The ESPRIT trial (Enhanced Suppression of the
Platelet IIb/Illa Receptor with Integrilin Therapy) was a placebo-controlled
trial designed to assess whether eptifibatide improved outcome
in patients undergoing stenting (14). Fourteen percent of the
2,064 patients enrolled in ESPRIT had UA/NSTEMI. The primary
end point (the composite of death, MI, target-vessel revascularization,
and "bailout" GP Ilb/Illa antagonist therapy) was
reduced from 10.5% to 6.6% with treatment (p = 0.0015). There
was consistency in the reduction of events in all components
of the end point and in all major subgroups, including patients
with UA/NSTEMI. Major bleeding occurred more frequently in patients
who received eptifibatide (1.3%) than in those who received
placebo (0.4%; p = 0.027); however, no significant difference
in thetransfusion rate occurred. At 1 year of follow-up, death
or MI occurred in 12.4% of patients assigned to placebo and
8.0% of eptifibatide-treated patients (hazard ratio 0.63; 95%
confidence interval [CI] 0.48 to 0.83; p = 0.001) (15).
In the only head-to-head comparison of GP Ilb/lila antagonists,
the TARGET trial (Do Tirofiban and ReoPro Give similar Efficacy?
Trial) randomized 5,308 patients to tirofiban or abciximab before
PCI with the intent to perform stenting (16). The primary end
point, a composite of death, nonfatal MI, and urgent target-vessel
revascularization at 30 days, occurred less frequently in those
given abciximab than in those given tirofiban (6.0% vs. 7.6%;
p = 0.038). There was a similar direction and magnitude for
each component of the end point. The difference in outcome between
the 2 treatment groups may be related to a suboptimal dose of
tirofiban resulting in inadequate platelet inhibition. However,
by six months, the primary end point occurred in a similar percentage
of patients in each group (14.9% tirofiban vs. 14.3 % abciximab,
NS). Mortality was also similar (1.9% vs. 1.7%, NS) (17). Glycoprotein
IIb/IIIa antagonists without scheduled PCI. The Global Utilization
of Strategies to Open Occluded Coronary Arteries IV-Acute Coronary
Syndromes (GUSTO IV-ACS) trial (18) enrolled 7,800 patients
with UA/NSTEMI who were admitted to the hospital with more than
5 min of chest pain and ST-segment depression and/or elevated
troponin T or I concentration and in whom early (less than 48
h) revascularization was not intended to be conducted. All received
ASA and either unfractionated heparin (UFH) or LMWH. They were
randomized to placebo, an abciximab bolus and 24-h infusion,
or an abciximab bolus and 48-h infusion. The primary end point,
death or MI at 30 days, occurred in 8.0% of patients given placebo,
8.2% given 24-h abciximab, and 9.1% given 48-h abciximab, differences
that were not statistically significant. At 48 h, death occurred
in 0.3%, 0.7%, and 0.9% in these groups, respectively (placebo
vs. abciximab 48 h, p = 0.008). The lack of benefit of abciximab
was observed in most subgroups, including patients with elevated
concentrations of troponin who were at higher risk. Although
the explanation for these results is not clear, they indicate
that abciximab, at least at the dosing regimen used in GUSTO
IV-ACS, is not indicated in the management of patients with
UA or NSTEMI in whom an early invasive management strategy is
not planned.
In the PRISM-PLUS trial, 1,069 patients did not undergo early
PCI. Although tirofiban treatment was associated with a lower
incidence of death, MI or death, and MI or refractory ischemia
at 30 days, these reductions were not statistically significant
(19). In a high-risk subgroup of these patients not undergoing
PCI (TIMI risk score greater than or equal to 4) (3), tirofiban
appeared to be beneficial whether they underwent PCI (odds ratio
[OR] 0.60, 95% CI 0.35 to 1.01) or not (OR 0.69, 95% CI 0.49
to 0.99). However, no benefit was observed in the patients at
lower risk (6). In the PURSUIT trial, eptifibatide reduced the
incidence of death or MI from 15.7% to 14.2% (RR 0.91; 95% CI
0.79 to 1.00; p = 0.032) (20).
Boersma et al performed a meta-analysis of
GP IIb/IIIa antagonists in all 6 large, randomized, placebo-controlled
trials, including GUSTO IV-ACS (18), which involved 31,402 patients
with UA/NSTEMI who were not routinely scheduled to undergo coronary
revascularization (21). A small reduction in the odds of death
or MI in the active treatment arm (11.8% vs 10.8%; OR 0.91,
95% CI 0.84 to 0.98; p = 0.015) was observed. Unexpectedly,
no benefit was observed in women (test for interaction between
treatment assignment and gender, p less than 0.0001). However,
women with positive troponins derived a treatment benefit that
was similar to men. In the meta-analysis, reductions in the
end points of death or nonfatal MI considered individually did
not achieve statistical significance.
Although not scheduled for coronary revascularization procedures,
11,965 of the 31,402 patients (38%) actually underwent PCI or
CABG within 30 days, and in this subgroup, the OR for death
or MI in patients assigned to GP IIb/IIIa antagonists was 0.89
(95% CI 0.80 to 0.98). In the other 19,416 patients who did
not undergo coronary revascularization, the OR for death or
MI in the GP IIb/IIIa group was 0.95 (95% CI 0.86 to 1.05, p
= NS). Major bleeding complications were increased in the GP
IIb/IIIa antagonist-treated group compared with those who received
placebo (1.4% vs. 2.4%, p less than 0.0001). The authors concluded
that in patients with UA/NSTEMI who were not routinely scheduled
for early revascularization and who were at high risk of thrombotic
complications, "treatment with a GP Ilb/Illa inhibitor
might therefore be considered" (21). Thus, GP Ilb/Illa
inhibitors are of benefit in high-risk patients with UA/NSTEMI,
and their administration, in addition to ASA and heparin, to
patients in whom catheterization and PCI are planned received
a Class I recommendation. These agents are of questionable benefit
in patients who do not undergo PCI. However, the revised guidelines
recommend broader indications for a routine invasive strategy
(see following text).
Thus, clopidogrel (in addition to aspirin and heparin or low
molecular weight heparin) is recommended for patients with UA/NSTEMI
in whom a noninterventional approach is planned (Class I recommendation).
In patients in whom an interventional approach is planned, a
GP IIb/IIIa inhibitor (in addition to aspirin and heparin or
low molecular weight heparin) is recommended (Class I recommendation).
No head-to-head comparison of clopidogrel, a GP IIb/IIIa inhibitor,
and their combination has been reported. The addition of a GP
IIb/IIIa inhibitor to a subset of patients in the CURE trial
who were receiving aspirin, clopidogrel, and heparin appeared
to be well tolerated, and current practice frequently involves
the use of this combination of drugs. However, until further
information on the safety and efficacy of such quadruple therapy
becomes available, a Class Ila recommendation is made for the
addition of a GP IIb/IIIa inhibitor for patients with UA/NSTEMI
who are transfusion rate occurred. At 1 year of follow-up, death
or MI occurred in 12.4% of patients assigned to placebo and
8.0% of eptifibatide-treated patients (hazard ratio 0.63; 95%
confidence interval [CI] 0.48 to 0.83; p = 0.001) (15).
In the only head-to-head comparison of GP Ilb/lila antagonists,
the TARGET trial (Do Tirofiban and ReoPro Give similar Efficacy?
Trial) randomized 5,308 patients to tirofiban or abciximab before
PCI with the intent to perform stenting (16). The primary end
point, a composite of death, nonfatal MI, and urgent target-vessel
revascularization at 30 days, occurred less frequently in those
given abciximab than in those given tirofiban (6.0% vs. 7.6%;
p = 0.038). There was a similar direction and magnitude for
each component of the end point. The difference in outcome between
the 2 treatment groups may be related to a suboptimal dose of
tirofiban resulting in inadequate platelet inhibition. However,
by six months, the primary end point occurred in a similar percentage
of patients in each group (14.9% tirofiban vs. 14.3 % abciximab,
NS). Mortality was also similar (1.9% vs. 1.7%, NS) (17). Glycoprotein
IIb/IIIa antagonists without scheduled PCI. The Global Utilization
of Strategies to Open Occluded Coronary Arteries IV-Acute Coronary
Syndromes (GUSTO IV-ACS) trial (18) enrolled 7,800 patients
with UA/NSTEMI who were admitted to the hospital with more than
5 min of chest pain and ST-segment depression and/or elevated
troponin T or I concentration and in whom early (less than 48
h) revascularization was not intended to be conducted. All received
ASA and either unfractionated heparin (UFH) or LMWH. They were
randomized to placebo, an abciximab bolus and 24-h infusion,
or an abciximab bolus and 48-h infusion. The primary end point,
death or MI at 30 days, occurred in 8.0% of patients given placebo,
8.2% given 24-h abciximab, and 9.1% given 48-h abciximab, differences
that were not statistically significant. At 48 h, death occurred
in 0.3%, 0.7%, and 0.9% in these groups, respectively (placebo
vs. abciximab 48 h, p = 0.008). The lack of benefit of abciximab
was observed in most subgroups, including patients with elevated
concentrations of troponin who were at higher risk. Although
the explanation for these results is not clear, they indicate
that abciximab, at least at the dosing regimen used in GUSTO
IV-ACS, is not indicated in the management of patients with
UA or NSTEMI in whom an early invasive management strategy is
not planned.
In the PRISM-PLUS trial, 1,069 patients did not undergo early
PCI. Although tirofiban treatment was associated with a lower
incidence of death, MI or death, and MI or refractory ischemia
at 30 days, these reductions were not statistically significant
(19). In a high-risk subgroup of these patients not undergoing
PCI (TIMI risk score greater than or equal to 4) (3), tirofiban
appeared to be beneficial whether they underwent PCI (odds ratio
[OR] 0.60, 95% CI 0.35 to 1.01) or not (OR 0.69, 95% CI 0.49
to 0.99). However, no benefit was observed in the patients at
lower risk (6). In the PURSUIT trial, eptifibatide reduced the
incidence of death or MI from 15.7% to 14.2% (RR 0.91; 95% CI
0.79 to 1.00; p = 0.032) (20).
Boersma et al performed a meta-analysis of GP IIb/IIIa antagonists
in all 6 large, randomized, placebo-controlled trials, including
GUSTO IV-ACS (18), which involved 31,402 patients with UA/NSTEMI
who were not routinely scheduled to undergo coronary revascularization
(21). A small reduction in the odds of death or MI in the active
treatment arm (11.8% vs 10.8%; OR 0.91, 95% CI 0.84 to 0.98;
p = 0.015) was observed. Unexpectedly, no benefit was observed
in women (test for interaction between treatment assignment
and gender, p less than 0.0001). However, women with positive
troponins derived a treatment benefit that was similar to men.
In the meta-analysis, reductions in the end points of death
or nonfatal MI considered individually did not achieve statistical
significance.
Although not scheduled for coronary revascularization
procedures, 11,965 of the 31,402 patients (38%) actually underwent
PCI or CABG within 30 days, and in this subgroup, the OR for
death or MI in patients assigned to GP IIb/IIIa antagonists
was 0.89 (95% CI 0.80 to 0.98). In the other 19,416 patients
who did not undergo coronary revascularization, the OR for death
or MI in the GP IIb/IIIa group was 0.95 (95% CI 0.86 to 1.05,
p = NS). Major bleeding complications were increased in the
GP IIb/IIIa antagonist-treated group compared with those who
received placebo (1.4% vs. 2.4%, p less than 0.0001). The authors
concluded that in patients with UA/NSTEMI who were not routinely
scheduled for early revascularization and who were at high risk
of thrombotic complications, "treatment with a GP Ilb/Illa
inhibitor might therefore be considered" (21). Thus, GP
Ilb/Illa inhibitors are of benefit in high-risk patients with
UA/NSTEMI, and their administration, in addition to ASA and
heparin, to patients in whom catheterization and PCI are planned
received a Class I recommendation. These agents are of questionable
benefit in patients who do not undergo PCI. However, the revised
guidelines recommend broader indications for a routine invasive
strategy (see following text).
Thus, clopidogrel (in addition to aspirin
and heparin or low molecular weight heparin) is recommended
for patients with UA/NSTEMI in whom a noninterventional approach
is planned (Class I recommendation). In patients in whom an
interventional approach is planned, a GP IIb/IIIa inhibitor
(in addition to aspirin and heparin or low molecular weight
heparin) is recommended (Class I recommendation). No head-to-head
comparison of clopidogrel, a GP IIb/IIIa inhibitor, and their
combination has been reported. The addition of a GP IIb/IIIa
inhibitor to a subset of patients in the CURE trial who were
receiving aspirin, clopidogrel, and heparin appeared to be well
tolerated, and current practice frequently involves the use
of this combination of drugs. However, until further information
on the safety and efficacy of such quadruple therapy becomes
available, a Class Ila recommendation is made for the addition
of a GP IIb/IIIa inhibitor for patients with UA/NSTEMI who are
receiving aspirin, clopidogrel, and unfractionated or low molecular
weight heparin and who are referred for an invasive strategy.
A Class I recommendation is made for a GP Ilb/Illa inhibitor
at the time of PCI in patients receiving heparin and aspirin.
Specific updated recommendations for the use of antiplatelet
regimens in the revised guidelines are as follows:
Class I
1. Antiplatelet therapy should be initiated
promptly. ASA should be administered as soon as possible after
presentation and continued indefinitely. (Level of Evidence:
A)
2. Clopidogrel should be administered to hospitalized patients
who are unable to take ASA because of hypersensitivity or major
gastrointestinal intolerance. (Level of Evidence: A)
*3. In hospitalized patients in whom an early noninterventional
approach is planned, clopidogrel should be added to ASA as soon
as possible on admission and administered for at least 1 month
(Level of Evidence: A), and for up to 9 months. (Level of Evidence:
B)
*4. A platelet GP IIb/IIIa antagonist should be administered,
in addition to ASA and heparin, to patients in whom catheterization
and PCI are planned. The GP IIb/IIIa antagonist may also be
administered just prior to PCI. (Level of Evidence: A)
*.5. In patients for whom a PCI is planned and who are not at
high risk for bleeding, clopidogrel should be started and continued
for at least 1 month (Level of Evidence: A) and for up to 9
months. (Level of Evidence: B)
*6. In patients taking clopidogrel in whom elective CABG is
planned, the drug should be withheld for 5 to 7 days. (Level
of Evidence: B)
Class Ila
*1. Eptifibatide or tirofiban should
be administered, in addition to ASA and LMWH or UFH, to patients
with continuing ischemia, an elevated troponin, or with other
high-risk features in whom an invasive management strategy is
not planned. (Level of Evidence: A)
*2. A platelet GP IIb/IIIa antagonist should be administered
to patients already receiving heparin, ASA, and clopidogrel
in whom catheterization and PCI are planned. The GP IIb/IIIa
antagonist may also be administered just prior to PCI. (Level
of Evidence: B)
Class IIb
*1. Eptifibatide or tirofiban, in addition
to ASA and LM" or UFH, to patients without continuing ischemia
who have no other high-risk features and in whom PCI is not
planned. (Level of Evidence: A)
Class III
1. Intravenous fibrinolytic therapy in
patients without acute ST-segment elevation, a true posterior
MI, or a presumed new left bundle-branch block. (Level of Evidence:
A)
*2. Abciximab administration in patients in whom PCI is not
planned. (Level of Evidence: A)
*New indication, not included in the September
2000 guidelines.
'Minor clarification different
from full-text version on web site.
Anticoagulant Therapy
The September 2000 guidelines (1) reviewed the evidence regarding
the use of intravenous UFH or subcutaneous LMWH. It provided
the following Class I recommendation:
"Parenteral anticoagulation with intravenous UFH or
subcutaneous LMWH should be added to antiplatelet
therapy with ASA or a thienopyridine. (bevel of Evidence: B)"
In the interim, a number of studies have appeared that support
the use of enoxaparin. In the EVET trial (Enoxaparin VErsus
Tinzaparin in the management of unstable coronary artery disease),
2 LMWHs, enoxaparin and tinzaparin, administered for 7 days,
were compared in 438 patients with UA/NSTEMI. A preliminary
report stated that both the recurrence of unstable angina and
the need for revascularization were significantly lower in the
enoxaparin group (22). Because the level of anticoagulant activity
cannot be easily measured in patients given LMWH (e.g., activated
partial thromboplastin time or activated clotting time), interventional
cardiologists have expressed concern about the substitution
of LMWH for UFH in patients scheduled for catheterization with
possible PCI. However, Collet et al. (23) have shown in a small
nonrandomized observation study in 293 patients that PCI can
be performed safely with UA/NSTEMI patients who received the
usual dose of enoxaparin. In NICE-1 (National Investigators
Collaborating on Enoxaparin), an observational study, intravenous
enoxaparin (1.0 mg/kg) was used in 828 patients undergoing elective
PCI without an intravenous GP IIb/ IIIa antagonist (24). The
rates of bleeding (L1% major bleeding and 6.2% minor bleeding
in 30 days) were comparable to those observed in historical
controls with UFH.
An alternative approach is to use LMWH during the period of
initial stabilization and to withhold the dose on the morning
of the procedure. If an intervention is required and more than
8 h has elapsed since the last dose of LMWH, UFH can be used
for PCI according to usual practice patterns. Because the anticoagulant
effect of UFH can be more readily reversed than that of LMWH,
UFH is preferred in patients likely to undergo CABG within 24h.
Class II
1. Intravenous fibrinolytic therapy in
patients without acute ST-segment elevation, a true posterior
MI, or a presumed new left bundle-branch block. (Level of Evidence:
A)
*2. Abciximab administration in patients in whom PCI is not
planned. (Level of Evidence: A)
JACC Vol. 40, No. 7, 2002 October 2, 2002:1366-74
The September 2000 guidelines reflected
concern regarding the combined use of LMWH and GP IIb/lila antagonists.
Although the data are not definitive, it now appears that GP
IIb/IIIa antagonists can be used with LMWH. In the ACUTE II
(Anti-thrombotic Combination Using Tirofiban and Enoxaparin
II) study (25), UFH and enoxaparin were compared in patients
with UA/NSTEMI who were given tirofiban. The frequencies of
both major and minor bleeding were similar, and there was a
trend to fewer adverse events in the patients given enoxaparin.
A number of other open-label studies have examined the safety
of combining enoxaparin with abciximab, eptifibatide, or tirofiban
in patients with UA/NSTEMI who are treated with PCI or conservatively;
of combining enoxaparin with abciximab in patients undergoing
elective PCI (26); and of combining dalteparin with abciximab
in patients with UAINSTEMI who are treated conservatively and
during PCI (27). Although the majority of these studies relied
on historical controls, none suggested that the combination
of enoxaparin and a GP Ilb/Illa antagonist was associated with
excess bleeding, whether or not the patient also underwent PCI.
Specific recommendations for the use of heparins in the revised
guidelines are as follows:
Class I
*1. Anticoagulation with subcutaneous
LMWH or intravenous UFH should be added to antiplatelet therapy
with ASA and/or clopidogrel. (Level of Evidence: A)
Class Ila
*fl. Enoxaparin is preferable to UFH
as an anticoagulant in patients with UA/NSTEMI, in the absence
of renal failure and unless CABG is planned within 24 h. (Level
of Evidence: A)
*New indication, not included in
the September 2000 guidelines.
*Minor clarification different from full-text version on web
site.
EARLY CONSERVATIVE VS. EARLY INVASIVE
STRATEGIES
The September 2000 guidelines indicated that
2 different treatment strategies, termed "early conservative"
and "early invasive," may be used in patients with
UA/NSTEMI (1). In the early conservative strategy, coronary
angiography is reserved for patients with evidence of recurrent
ischemia (angina at rest or with minimal activity or dynamic
STsegment changes) or a strongly positive stress test despite
vigorous medical therapy. In the early invasive strategy, patients
without clinically obvious contraindications to coronary revascularization
are routinely recommended for coronary angiography and angiographically
directed revascularization, if possible.
Several trials comparing these 2 strategies
were reviewed,but greatest attention was paid to the then-most-recent
trial, FRISC II (Fragmin and Fast Revascularization during InStability
in Coronary artery disease II) (28). At 1 year, the mortality
rate in the invasive strategy group was 2.2% compared with 3.9%
in the noninvasive strategy group (p = 0.016) (29). However,
in FRISC II, the invasive strategy involved treatment for an
average of 6 days in the hospital with LMWH, ASA, nitrates,
and beta-blockers before coronary angiography, an approach that
would be difficult to adopt in U.S. hospitals.
In the interim, the TACTICS-TIMI 18 trial was reported (7).
In this trial, 2,220 patients with UA or NSTEMI were treated
with ASA, heparin, and the GP IIb/IIIa antagonist tirofiban.
They were then randomized to an early invasive strategy with
routine coronary angiography within 48 h followed by revascularization
if the coronary anatomy was deemed suitable, or to a more conservative
strategy. In the latter, catheterization was performed only
if the patient had recurrent ischemia or a strongly positive
stress test. Death, myocardial (re)infarction, or rehospitalization
for an acute coronary syndrome at 6 months occurred in 19.4%
of patients assigned to the conservative strategy vs. 15.9%
assigned to the invasive strategy (OR 0.78; 95% CI 0.62 to 0.97;
p = 0.025). Occurrence of death or MI was also reduced at 6
months (9.5 % vs 7.3%; p less than 0.05). The beneficial effects
on outcome were particularly evident in medium- and high-risk
patients, as defined by an elevation of troponin T greater than
0.01 ng/ml or of troponin I greater than 0.1 ng/ml, the presence
of STsegment deviation, or a TIMI risk score greater than or
equal to 3 (7,30). In the absence of these high-risk features,
outcomes in patients assigned to the 2 strategies were similar.
Rates of major bleeding were similar, and lengths of hospital
stay were reduced in patients assigned to the invasive strategy.
The benefits of the invasive strategy were achieved at no significant
increase in the cost of care over the 6-month follow-up period.
Thus, both the FRISC 11 (28,29) and TACTICS-TIMI 18 (7,30) trials,
the 2 most recent trials comparing invasive vs. conservative
strategies in patients with UA/NSTEMI, showed a benefit in patients
assigned to the invasive strategy. In contrast to earlier trials,
a large majority of patients undergoing PCI in these 2 trials
received coronary stents as opposed to balloon angioplasty alone.
In TACTICS-TIMI 18, treatment included the GP IIb/IIIa antagonist
tirofiban, which was administered for an average of 22 h before
coronary angiography. The routine use of the GP IIb/IIIa antagonist
in this trial may have eliminated the excess risk of early (within
7 days) acute MI in the invasive arm, an excess risk that was
observed in FRISC II and other trials in which there was no
routine "upstream" use of a GP Ilb/Illa antagonist.
Therefore, an invasive strategy is associated with a better
outcome in UA/NSTEMI patients at high risk who receive a GP
Ilb/Illa antagonist (7). Although the benefit of GP Ilb/Illa
antagonists is well established for patients with UA/NSTEMI
who undergo PCI, the optimumtime of commencing these drugs-as
early as possible after presentation, i.e. "upstream,"
as in TACTICS-TIMI 18, or just before the PCI-has not been established.
Specific recommendations for the use of an invasive strategy
in the revised guidelines are as follows:
Class I
*1. An early invasive strategy in patients
with UA/ NSTEMI without serious comorbidity and who have any
of the following high-risk indicators:
(Level of Evidence: A)
*a) Recurrent angina/ischemia at rest or with lowlevel activities
despite intensive anti-ischemic therapy.
*b) Elevated TnT or TnI
*c) New or presumably new ST-segment depression
d) Recurrent angina/ischemia with CHF symptoms, an S3 gallop,
pulmonary edema, worsening rales, or new or worsening MR
e) High-risk findings on noninvasive stress testing
f) Depressed LV systolic function (e.g., EF less
than 0.40 on noninvasive study) g) Hemodynamic instability
h) Sustained ventricular tachycardia i) PCI within 6 months
j) Prior CABG
2. In the absence of any of these findings, either an early
conservative or an early invasive strategy may be offered in
hospitalized patients without contraindications for revascularization.
(Level of Evidence: B)
*New indication, not included in the September 2000
guidelines.
*Minor clarification different from full-text version on web
site.
RISK FACTOR MODIFICATION
The September 2000 guidelines pointed
out that despite the overwhelming evidence for the benefits
of beta-hydroxybeta-methylglutaryl-coenzyme A (HMG-CoA) reductase
(statin) therapy in patients with elevated low-density lipoprotein
(LDL) cholesterol levels, almost no data existed about the timing
of initiation of therapy in patients with acute coronary syndromes
(1). Indeed, the secondary prevention trials of statins specifically
excluded patients with UA/NSTEMI in the acute phase. Fewer than
300 patients had been entered into the trials within 4 months
of an acute coronary syndrome.
The Lipid-Coronary Artery Disease (L-CAD) study was a small
trial that randomized 126 patients with an acute coronary syndrome
to early treatment with pravastatin, alone or in combination
with cholestyramine or niacin, or to usual care. At 24 months,
the patients who received early aggressive treatment had a lower
incidence of clinical events
(23%) than the usual-care group (52%; p = 0.005) (31). In the
MIRACL (Myocardial Ischemia Reduction with Aggressive Cholesterol
Lowering) trial, 3,086 patients were randomized to treatment
with an aggressive lipid-lowering regimen of atorvastatin 80
mg per day or placebo 24 to 96 h after an acute coronary syndrome
(32). At 16 weeks of follow-up, the primary end point of death,
nonfatal MI, resuscitated cardiac arrest, or recurrent severe
myocardial ischemia was reduced from 17.4% in the placebo group
to 14.8% in the atorvastatin group (p = 0.048). There were no
significant differences between the 2 groups in the risk of
the following individual end points: death, nonfatal MI, cardiac
arrest, or worsening heart failure; however, there were fewer
strokes and a lower risk of severe recurrent ischemia in patients
assigned to atorvastatin.
Although the evidence from these 2 trials of a beneficial effect
of predischarge initiation of lipid-lowering therapy is not
yet robust or definitive, observational studies support this
policy. In the Swedish Registry of Cardiac Intensive Care of
almost 20,000 patients, the adjusted relative risk of mortality
was 25% lower in patients in whom statin therapy was initiated
before hospital discharge (33). In addition, patients in whom
lipid-lowering therapy is begun in the hospital are much more
likely to be undergoing such therapy at a later time. In one
demonstration project, the Cardiovascular Hospitalization Atherosclerosis
Management Program (CHAMP), the in-hospital initiation of lipidlowering
therapy was associated with an increased percentage of patients
treated with statins 1 year later (from 10% to 91%) and with
a higher frequency of patients whose LDL cholesterol was less
than 100 mg/dl (from 6% to 58%) (34). Although additional trials
are ongoing, there appear to be no adverse effects and substantial
advantages to the initiation of lipid-lowering therapy before
hospital discharge (35-37). Such early initiation of therapy
has also been recommended in the third report of the National
Cholesterol Education Program (NCEP III), which also raised
the threshold of high-density lipoprotein cholesterol concentration
that required therapy (38). Similar considerations apply to
the early initiation of statin therapy following PCI. In the
Lescol Intervention Prevention Study (LIPS), 1,669 patients
were randomized to receive 80 mg fluvastatin or placebo, beginning
two days after PCI. After a follow-up of 3.9 years, the statin-treated
group had a lower incidence of clinical events (21.4%) than
the placebo group (26.7%), p = 0.01 (39).
In addition to maintaining the original Class I recommendations
for LDL cholesterol reduction, specific additional recommendations
for the use of lipid-lowering therapy in UA/NSTEMI in the revised
guidelines are as follows:
Class I
*1. A fibrate or niacin if high-density
lipoprotein cholesterol is less than 40 mg per dl, occurring
asan isolated finding or in combination with other lipid abnormalities.
(Level of Evidence: B)
Class Ila
*1. HMG-CoA reductase inhibitors and
diet for LDL cholesterol greater than 100 mg per dl begun 24
to 96 h after admission and continued at hospital discharge.
(Level of Evidence: B)
*New indication, not included in
the September 2000 guidelines.
CONCLUSIONS
These guidelines address the diagnosis and
management of patients with UA and the closely related condition
NSTEMI. These life-threatening disorders are a major cause of
emergency medical care and are responsible for more than 1.4
million hospitalizations annually in the United States (40).
Nearly 60% of these admissions are among persons greater than
65 years old, and almost half occur in women. In 1997, there
were 5,315,000 visits to US emergency departments for the evaluation
of chest pain and related symptoms (41).
Because of the high incidence of UA/NSTEMI
and the seriousness of this condition (approximately 15% rate
of death or [re]infarction at 30 days) (1,20), continued research
in this field is of the greatest importance. It is encouraging
that in the 21 months since the publication of the September
2000 guidelines, a considerable body of additional useful information
about these conditions has emerged. Indeed, the progress between
September 2000 and June 2002 equals that between 1994, when
the first guidelines were published (42), and September 2000.
Editorial:N Engl Med,Vol.347,No.13,September26,2002,Pp
1019-1021
ANTITHROMBOTIC THERAPY AFTER MYOCARDIAL
INFARCTION
DESPITE encouraging trends over the past three decades, coronary
heart disease remains the leading cause of death in the United
States and other industrialized countries. Recent data from
the National Center for Health Statistics and the National Heart,
Lung, and Blood Institute emphasize the full dimensions of this
health problem, revealing that nearly 13 million Americans have
coronary heart disease and that 7.5 million have had a myocardial
infarction. Because there are 1.1 million myocardial infarctions
in the United States alone each year and because 450,000 of
them represent recurrent infarctions, which carry an inherently
greater risk of death and disability than first events, the
importance of secondary-prevention strategies that can be widely
implemented is unparalleled in health care.
In this issue of the Journal, the potential role of oral anticoagulant
therapy as secondary prevention is highlighted in the Warfarin,
Aspirin, Reinfarction Study (WARIS II), reported by Hurlen et
al. In this open-label study, patients hospitalized in 20 Norwegian
centers for acute myocardial infarction were randomly assigned
to long-term treatment with either warfarin (at a dose targeted
to achieve an international normalized ratio [INR] of 2.8 to
4.2), 160 mg of aspirin daily, or 75 mg of aspirin daily plus
warfarin (INR, 2.0 to 2.5). The primary outcome - a composite
of death, nonfatal reinfarction, or thromboembolic stroke -
occurred in 20.0 percent of the patients in the aspirin-only
group, 16.7 percent of those in the warfarin-only group, and
15.0 percent of those in the combination-therapy group. The
overall risk reduction was 29 percent in the combination-therapy
group (P=0.001 for the comparison with the aspirin-only group)
and 19 percent in the warfarin-only group (P=0.03). The incidence
of nonfatal major hemorrhage among the patients receiving warfarin
(either alone or in combination) was three to four times that
among the patients receiving aspirin alone.
Does the hypothesis that oral anticoagulant therapy may be effective
for secondary prevention after myocardial infarction have a
sound scientific basis? Angioscopic studies have documented
residual thrombosis and soft (vulnerable) plaques in the majority
of patients for several months after myocardial infarction,
with coexisting evidence of thrombin generation. Thrombin that
is incorporated into plasma fibrin clots, and its subsequent
binding to purified fibrin, is saturable and reversible. Thrombin
activity, determined by in situ zymography, is approximately
4.5 IU per gram (wet weight) of thrombus . Although there is
firm evidence that thrombin is the primary procoagulant enzyme
in both physiologic hemostasis and pathologic thrombosis, its
mechanism of generation and regulatory functions represent critical
considerations in the development of effective therapies for
cardiovascular thrombotic disorders.
A cell-based model of vascular thrombosis, which fosters a functional,
physiologic view of complex biochemical events on cell surfaces,
identifies tissue factor-bearing cells (monocytes and endothelial
cells) as the initiating sites of coagulation (Fig. 1). The
complexing of tissue factor with factor Vila (from plasma) leads
to thrombin generation, which in turn activates platelets by
way of protease-activated receptors. The final phase takes place
on platelet surfaces after the assembly of tenase complex (factor
Villa and tissue factor-factor VIIa complex) and prothrombinase
complex (factors Va and Xa, calcium, and phospholipid).
The recognized contribution of inflammation to atherothrombosis
also underscores the importance of coagulation proteases and
thrombin generation on nonplatelet surfaces. Inflammatory cytokines,
including tumor necrosis factor a and interleukin-1, facilitate
thrombin generation by stimulating the release of tissue factor
from monocytes and vascular endothelial cells. In addition,
they impair fibrinolysis through the provoked release of thrombin-activatable
fibrinolysis inhibitor and plasminogen-activator inhibitor type
1. Inflammatory cytokines, by reducing the concentration of
endothelial-cell-surface thrombomodulin and the formation of
thrombomodulin-activated protein C complex, compromise thromboresistance
to factors Va and VIIla. Lastly, leukocytes adhered to activated
endothelial cells by P-selectin glycoprotein ligand 1 in regions
of variable shear stress7 promote thrombosis by several unique
pathways, such as the de-encryption of tissue factor, the binding
of macrophage antigen 1 (CD11b-CD18) to coagulation factor Xa,
and the capture of fibrin protofibrils.
It is the direct involvement of coagulation proteases in thrombotic,
inflammatory, and cellular regulatory processes that provides
a scientific underpinning to the consideration of anticoagulant
agents in secondary prevention. The four hydroxycoumarin compounds
used currently in clinical practice - warfarin, phenprocoumon,
acenocoumarol, and dicumarol - inhibit the vitamin K-dependent,
post-translational carboxylation of coagulation factors II,
VII, IX, and X (which is required for calcium-mediated binding
of these factors to the negatively charged phospholipids found
in platelets and injured endothelial cells). The ability to
inhibit thrombin generation offers consid
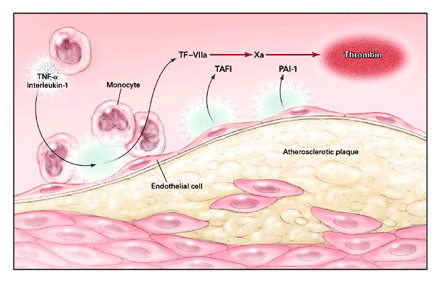 Figure
1.
Figure
1.
A Cell-Based Paradigm of Arterial Thrombosis.
Arterial thrombosis, though traditionally
viewed as a platelet-dependent process, occurs on a variety
of other cell surfaces, including the surfaces of activated
endothelial cells and monocytes, which are tissue factor (TF)-bearing
cells. Inflammatory cytokines (tumor necrosis factor a [TNF-a]
and interleukin-1) impair surface fibrinolysis by the provoked
release of thrombin-activatable fibrinolysis inhibitor (TAFI)
and plasminogen-activator inhibitor type 1 (PAT-1). Oral anticoagulants
potentially interrupt several prothrombotic, inflammatory, and
regulatory processes.
erable appeal; however, inhibition of one or more specific coagulation
proteases may also prove beneficial. Tissue factor requires
a cofactor, factor VIIa from plasma, to fulfill its enzymatic
capabilities. Factor Xa, a vital component of prothrombinase-mediated
conversion of prothrombin to thrombin, induces the expression
of tissue factor from endothelial cells, smooth-muscle cells,
and macrophages; increases endothelial-cell expression of E-selectin,
intercellular adhesion molecule 1, and vascular-cell adhesion
molecule 1, with subsequent leukocyte adhesion; and stimulates
the synthesis and release of interleukin-6, interleukin-8, and
monocyte chemotactic protein 1.9 Thus, our knowledge of atherosclerosis,
inflammation, and thrombosis firmly supports a hypothesis designed
to test oral anticoagulant therapy for the secondary prevention
of cardiovascular events after myocardial infarction.
The evolution of oral anticoagulant agents for the management
of acute coronary syndromes has taken a circuitous path, although
much insight has been achieved along the way. In WARIS I,10
patients with myocardial infarction received either warfarin
(INR, 2.8 to 4.8) or placebo. With warfarin, the incidence of
major hemorrhage was twice that with placebo, but mortality
and the rate of reinfarction were reduced by 24 percent and
34 percent, respectively. Interest in oral anticoagulant therapy
then waned during the 1990s because two large-scale trials,
the Coumadin Aspirin Reinfarction Study (CARS) (median INR,
1.3)" and the Combination Hemotherapy and Mortality Prevention
(CHAMP) study (median INR, 1.8)12 found no reduction in mortality,
in the rate of reinfarction, or in the rate of stroke with warfarin
(alone or in combination with aspirin) as compared with aspirin
monotherapy.
The favorable results observed in WARTS I, coupled with the
disappointing findings of CARS and CHAMP, not only established
the need for a definitive trial of anticoagulant therapy in
acute coronary syndromes but also raised the important possibility
of a "threshold" level of anticoagulation for benefit
(as previously observed in venous thromboembolic disorders and
atrial fibrillation). Indeed, WARTS II,2 the recently published
Antithrombotics in the Secondary Prevention of Events in Coronary
Thrombosis 2 study, 13 and the Antithrombotics in the Prevention
of Reocclusion in Coronary Thrombolysis 2 trial 14 support a
target level of anticoagulation approaching an INR of 3.0 (range,
2.5 to 3.5) for anticoagulation monotherapy and of 2.5 (range,
2.0 to 3.0) for combination therapy with aspirin. Thus, the
available data, based on nearly 20,000 patients participating
in randomized clinical trials, are strong and show that oral
anticoagulants, when given in adequate doses, reduce the rates
of reinfarction and thromboembolic stroke but at the cost of
increased rates of hemorrhagic events.
Maximizing the benefit associated with oral anticoagulant therapy
while minimizing the risk is a key consideration in management
strategies designed to achieve and maintain a target level of
inhibition. Because coumarin compounds have complex pharmacokinetic
and pharmacodynamic properties and are among the most challenging
drugs to regulate, coordinated anticoagulation clinics may be
the preferred means to provide safe and effective care. Accumulating
data show a 50 percent reduction in the rate of thromboembolism,
major hemorrhage, and emergency medical visits with the use
of this strategy; the use of portable, point-of-care coagulation
monitors, by allowing frequent testing, may improve outcomes
further." Even under ideal circumstances, the complexities
of coumarin therapy create real obstacles for clinicians and
their patients. In WARTS II, the INR in approximately one third
of the patients receiving warfarin alone was below the target
range; one third discontinued warfarin treatment at some point
during the 80-month study period; 5 to 7 percent were withdrawn
from treatment because of hemorrhagic complications; and 2 to
3 percent were deemed noncompliant. The exclusion of patients
75 years of age or older undoubtedly reduced the warfarin-associated
risk of hemorrhage.
In the United States, percutaneous coronary intervention and
stenting are performed in 15 to 20 percent of patients who have
myocardial infarction with ST-segment elevation, and an additional
20 to 40 percent undergo percutaneous coronary intervention
within the subsequent six weeks. The proportion of patients
who have myocardial infarction without ST-segment elevation
and who undergo percutaneous coronary intervention is even higher.
Histologic examination reveals platelets and leukocytes clustered
around the stent struts, and electron microscopy identifies
fibrinogen, prothrombin, thrombin, and tissue factor on the
outer surface of the platelet monolayer. Therapies designed
primarily to inhibit platelet activation and aggregation take
existing pathobiologic processes into consideration and are
preferred to anticoagulant therapy in this setting.
The contribution of WARTS II to our current knowledge base is
multidimensional. First, targeted inhibition of one or more
coagulation proteases (and, potentially, of their proinflammatory
or cell-regulatory properties) represents an important physiological
concept in arterial thrombosis. Second, a threshold level of
inhibition is required for benefit:. Third, close monitoring
and meticulous dose adjustment are absolute prerequisites for
safe and effective treatment. Fourth, oral anticoagulant agents
with .more predictable pharmacokinetic and pharmacodynamic properties
than coumarin compounds and with a broader therapeutic index
should be developed.
Although it is likely that antiplatelet therapy will remain
the standard of care in many countries for secondary prevention
after myocardial infarction, the findings of WARTS II must not
be overlooked. Oral anticoagulant therapy should be strongly
considered for patients at risk for thromboembolic events, those
with thrombophilia involving the arterial system (e.g., the
antiphospholipid syndrome), and - pending further investigation
of combination antiplatelet therapy in acute coronary syndromes
associated with ST-segment elevation - patients with possible
aspirin resistance. Advances in pharmacogenomics will ultimately
permit patient-specific antithrombotic therapy for patients
with acute coronary syndromes and other thrombotic disorders.
RICHARD C. BECKER, M.D.
University of Massachusetts Medical School Worcester, MA 01605
Editor's note: Dr. Becker receives research support from the
National Heart, Lung, and Blood Institute, Daiichi Pharmaceuticals,
and Merck, and he has received speaking fees from Aventis.
REFERENCES
1. Morbidity & mortality: 2002 chart book
on cardiovascular, lung, and blood diseases. Bethesda, Md.:
National Heart, Lung, and Blood Institute, May 2002.
2. Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin,
aspirin, or both after myocardial infarction. N Engl J Med 2002;347:96974.
3. Ueda Y, Asakura M, Yamaguchi 0, Hirayama A, Hori M, Kodama
K. The healing process of infarct-related plaques: insights
from 18 months of serial angioscopic follow-up. J Am Coll Cardiol
2002;38:1916-22. 4. Mutch NJ, Robbie LA, Booth NA. Human thrombi
contain an abundance of active thrombin. Thromb Haemost 2001;86:1028-34.
5. Nelken NA, Soifer SJ, O'Keefe J, Vu TK, Charo IF, Coughlin
SR. Thrombin receptor expression in normal and atherosclerotic
human arteries. J Clin Invest 1992;90:1614-21.
6. Hoffman M, Monroes DM III. A cell-based model of hemostasis.
Thromb Haemost 2001;85:958-65.