

Cardiomyopathy
Cardiomyopathy represents a group of diseases of the heart, which involve the heart muscle itself resulting in contractile and relaxation dysfunction of both ventricles leading to progressive chamber dilatation and then hypocontractile walls.
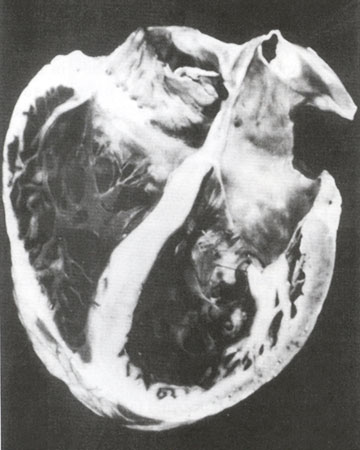
Fig. 43b
Gross pathology of a dilated cardiomyopathy depicting biventricular and biatrial dilatation and enlargement. Note the thinning of both the right ventricular and left ventricular free walls and the significant increase in ventricular volumes.
Hess, M.L., Pathak, S.K., Dilated Cardiomyopathies, Hurst's The Heart Update I, Chapter 6, p 125
Causes of Cardiomyopathy
A. Genetic Abnormalities
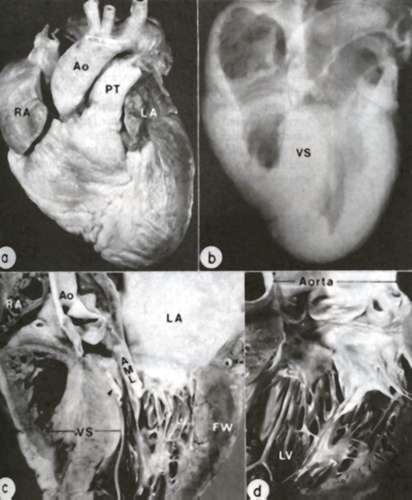
Figure 39a
Anatomic features of HCM are demonstrated in the heart of a 26-year-old man. A. Exterior view; both right atrium (RA) and left atrium (LA) are dilated. Ao= aorta; PT= pulmonary trunk. B. Radiograph of specimen showing asymmetric thickening of ventricular septum (VS). C. Coronal section ; the septum is clearly thicker than left ventricular free wall (FW); an endocardial mural contact plaque (arrow) is present in the left ventricular outflow tract in apposition to the anterior mitral leaflet (AML). D. closer view of plaque and thickened anterior leaflet.
(From WC Roberts, VJ Ferrans: Hum Pathol 6:287-342, 1975.)
1. Idiopathic hypertrophic cardiomyopathy (see more: fig.40a, fig.40b, fig.40c, fig.40d, fig.40e, fig.40f,, fig.41, fig.61 )
a. Age mainly young people
b. Abnormally thick IVS (see fig.39a, fig.39b, fig.39f, fig.40a, fig.40b, fig.40c, fig.40d, fig.40e, fig.40f, fig.41,)
c. Abnormal tissue staining properties (see fig.39c, fig.39d)
d. EKG abnormalities ( see fig.61 )
B. Alcoholism
Cardiomyopathy
DEFINITION/DIAGNOSIS
An alcohol cardiomyopathy is said to
be present when other causes of a dilated cardiomyopathy have
been excluded and there is a history of heavy, sustained alcohol
intake. The usual requirement in terms of alcohol amount is
100 g alcohol per day, typically over several years. However,
in susceptible individuals it is likely that lower amounts of
intake can produce a cardiomyopathy. The histologic features
of alcohol cardiomyopathy are nonspecific and do not differ
from IDC. Other than history, the only potentially distinguishing
feature between IDC and alcohol cardiomyopathy is that the latter
may present with a relatively high cardiac output.
DISTINCT PATHOPHYSIOLOGY
The pathophysiology of alcohol cardiomyopathy
is thought to he related to the toxic effects of alcohol, plus
in some subjects nutritional components such as thiamine deficiency.
PROGNOSIS
The prognosis depends on the degree of
impairment of myocardial function and the extent of abstinence
from alcohol and, in an extremely compromised patient, the administration
of thiamine. There is evidence that the prognosis is somewhat
better for alcohol cardiomyopathy than for IDC.
TREATMENT
The treatment of alcohol cardiomyopathy
does not differ from IDC, except the inclusion of total abstinence
from alcohol. Obviously, these subjects are not good candidates
for cardiac transplantation because of the high relapse rate
to alcoholism.
C. Adramycin, Daunorubicin, Doxorubicin Toxicity
(used in chemotheraphy for cancer )
D. Viral Infections
1. Coxsachie B
2. HIV
E. Chagas' Disease (Parasitic)
Chagas' Cardiomyopathy
DEFINITION/DIAGNOSIS
Chagas' disease is discussed as a cause of myocarditis. In addition, Chagas' disease is the most common cause of nonischemic cardiomyopathy in South and Central America, with over 10 million people afflicted. It is caused by a parasite, the leishmanial or tissue form of the protozoan Trypanosoma cruzi.
In settings as above where most transmissions are vectorborne, triatomine bugs defecate and inoculate trypomastigotes of the protozoan T.cruzi at mucosal surfaces or wound(bite sites).
Although in the United States the vector (triatoma, or kissing bug) is found only in the Southwest, Chagas' disease may be transmitted by blood transfusions, and as a result, it could become relatively more important in this country.
Additonal modes of transmissions have been described, including oral and vertical(transplacential) routes and organ donations. Transmissions by means of transfusions and organ donations are of growing significance in the United States and other industralied countries. As many as 20- 40% of infected persons will enter the chronic phase and have complications,primarily cardiac or gastrointestinal. Immunosuppress hosts can also have systemic parasitemia on reactivation of infection or neurologic manifestions including menigencephalitis.
The natural history consists of an initial myocarditis most commonly presenting in childhood, associated with acute myocardial infection followed by recovery and in some individuals the development of a dilated cardiomyopathy 10 to 30 years later.
In the acute phase, the diagnosis of Chagas' disease is made by visualization of parasites on thick and thin blood smears stained with Giemsa, but parasites become difficult to detect by 3 months after the infection.
The diagnosis of Chagas' cardiomyopathy is based on clinical (history, LV function, and electrocardiographic) criteria and a positive serologic test for T. cruzi.
Two tests are routinely run routinely performed: an enzyme-linked immunosorbent assay(ELISA) and an indirect immunoflourescence test for antobodies. Although the indirect immunoflourescence test is quite sensitive, cross-reactivity can be seeen with serum samples from patients with leishmaniasis.
Electrocardiographic abnormalities consist of bundle-branch or hemiblocks (indeed, hemiblocks were first described by Rosenbaum et al) in Chagas' afflicted hearts with discrete foci of involvement), LV hypertrophy, and first- or second-degree atrioventricular (AV) block.
The histologic lesion of chronic Chagas' consists of mononuclear infiltrates, fibrosis, and as shown in Fig. 74f below foci of the leishmanial form of T. cruzi in myocardial fibers.
The LV functional abnormalities initially may be segmental and may include an apical aneurysm but later become more global.
FIGURE
74f. Leishmanial forms of T.
cruzi within the swollen cytoplasm of a cardiac myocyte (Chagas'
&E stain, x250).
(Courtesy Dr. Elmer Koneman.)
DISTINCT PATHOPHYSIOLOGY
The basis for Chagas' cardiomyopathy is unknown but may be immunologic, whereby antibodies generated against T. cruzi crossreact with cardiac "
PROGNOSIS
The prognosis is relatively good
for a dilated cardiomyopathy and similar toIDC; the five-year
survival with heart failure is around 50 percent. Compared with
IDC, death likely occurs more commonly due to an arrhythmin
mechanisn. However. as for IDC and most othert dilated cardiomyopathies,
mortality risk depends directly o the degrees of ventricular
dysfunction and exercise intolerance.
TREATMENT
There is no definitive treatment for
Chagas' cardiomyopathy, and nonspecific treatment includes pacemaker
implantation for heart block and heart failure treatment as
for IDC. The one exception may be the more frequent use of amiodarone,
which appears to be particularly effective in treating arrhythmias
associated with Chagas' cardiomyopathy and in one study reduced
mortality compared with standard treatment. The role of cardiac
transplantation is still somewhat uncertain, but it can be done
at acceptable risk, especially when coupled with trypanocidal
SUMMARY
Dilated cardiomyopathies are important
because they are the most common cause of heart failure, which
is the single most costly medical problem in the adult U.S.
population. Cardiomyopathies in general are a heterogeneous
group of diseases, but they can be classified under a newly
modified WHO/ISFC classification system, which, although imperfect,
should he of great value in standardizing the terminology and
encouraging systematic investigative and clinical approaches
to diagnosis and treatment. Within this classification system,
primary and secondary dilated cardiomyopathies comprise the
single largest and most important group. Current diagnosis and
treatment of dilated cardiomyopathies vary somewhat among the
various types, but the cornerstones of medical management are
similar in most cases.
Genetic causes and influences on the natural history of dilated
cardiomyopathies are the new frontier in this field, and their
elucidation is almost certain to lead to new therapeutic and
diagnostic approaches. In the near future, molecular genetic
testing will be done routinely for many cardiomyopathies that
may have a single gene defect as the cause. As we learn more
about the influence of polymorphic genetic variation on the
natural history and selection of specific medical therapy, genetic
testing will be performed in most patients with cardiomyopathy.
F. Bacterial
G. Fungal
H. Amyloidosis
(see fig.43a, fig.73a, fig.74a, fig.76, fig.77a)
I. Hemchromatosis
(Abnormal deposition of iron in heart muscle)
J. Ischemic Coronary Artery Disease
(see fig.53, fig.54, fig.55, fig.56a, fig.70)
K. Arrhythmogenic RV Dysplasia
FAMILIAL ARRHYTHMOGENIC RIGHT
VENTRICULAR DYSPLASIA (ARVD)
Arrhythmogenic right ventricular
dysplasia is characterized by fatty infiltration of the right
ventricle, fibrosis, and ultimately thinning of the wall with
chamber dilatation. It is the most common cause of sudden cardiac
death in the young in Italy and is said to account for about
17 percent of sudden death in the young in the United States.
Rampazzo et al. mapped this disease in two families, one to
1q42-q43, and the other on chromosome 2q32; a third locus was
mapped to 14q12. A large Greek family with arrhythmogenic right
ventricular dysplasia and Naxos diseasewas recently mapped to
17q. Two loci responsible for AR\'D in North America were recently
mapped at 3p23 and the other at lOpl2. This is a very devastating
disease, since the first symptom is often sudden death. Electrocardiographic
abnormalities include inverted T waves in the right precordial
leads, late potentials, and right ventricular arrhythmias with
left bundle-branch block (LBBB).This is compounded by me great
difficulty in making tne diagnosis even when the condition occurs
in a family with a history of the disease. Since the disease
affects only the right ventricle, it is difficult to detect.
There is no definitive diagnostic standard. The right ventricular
biopsy is definitive when positive but often produces a false-negative
result, since the disease initiates in the epicardium and spreads
to the endocardium of the right ventricular free wall, making
it
inaccessible to biopsy. Consensus diagnostic criteria was developed
that include right ventricular biopsy, magnetic resonance imaging
(MRI), echo-cardiography, and electrocardiography. Identification
of the gene will have tremendous diagnostic impact and hopefully
will provide an explanation as to why ARVD is restricted to
the right ventricle. Is it a specific right ventricular chamber
gene? Is there a stimulus that is unique or predominates in
the right ventricle that precipitates the phenotype? What is
the stimulus? There are data suggesting that apoptosis is the
process leading to the development of fat and fibrosis in ARVD.
Discovery of a gene should shed light on the apoptosis pathway.
Arrhythmogenic right ventricular dysplasia
(ARVD) is predominantly right ventricular cardiomyopathy characterized
by fatty or fibrofatty replacement of myocardium. It is a rare
cause of sudden cardiac death except in a few endemic regions.
Recurrent ventricular tachycardia with multiple left bundle-branch
block morphologies typifies this disorder. It is a familial
disorder in approximately 30 percent of cases, with an autosomal
dominant mode of inheritance. The gene defect has been localized
to chromosomes 1, 3, and 14 In the fibrofatty variety, patchy
myocarditis, programmed cell death, and/or congenital abnormalities
of development appear to lead to myocardial atrophy and repair
by fibrofatty replacement, which may become the basis for reentrant
ventricular arrhythmia. The left ventricle and ventricular septum
can be involved in 50 to 67 percent of cases, especially later
in the course of the disease, and such involvement confers a
poor prognosis.
The electrocardiographic manifestations in sinus rhythm include
T-wave inversion in V1—V3 or complete or incomplete right
bundle-branch block. Intraventricular conduction delay may produce
a terminal notch on the QRS complex called an epsilon wave in
approximately 50 percent of patients. The ventricular ectopy
is usually of a left bundle-branch pattern, with a QRS axis
between —90° and +110°, and generally arises from
one of three sites of fatty degeneration. Called the triangle
of dysplasia, these sites are the right ventricular outflow
and inflow tract and apex. Any patient with frequent premature
beats of a left bundle-branch morphology and left-axis deviation
should be evaluated for this disorder.
In patients with ARVD, particularly at early stages of the disease,
ventricular tachycardia is often precipitated by exercise, and
its induction is usually found to be catecholamine sensitive
at electrophysiologic study. The course and prognosis of ARVD
are highly variable and difficult to predict. The annual incidence
of sudden cardiac death in ARVD has been estimated to be about
2 percent despite various treatments.
Righe ventricular dysplasiais a cardiomyopathy predominantly
of the right ventricle. Left ventricle involvement is usually
of a lesser and variable degree. Several anomalies may be included
under this general heading: Uhi’s anomaly, arrhythmogenic
right ventricular dysplasia, and right ventricular cardiomyopathy.
It is currently recognized as an important inherited cardiomyopathy
and a cause of sudden death, especially in youth. Its cause
is unknown, although an autosomal dominant pattern with variable
expression and penetrance has been suggested, since many cases
show a strong familial tendency.
Clinically patients typically present with recurrent ventricular
tachycardia of left-bundle-branch-block morphology and, less
commonly, CHF. Standard electrocardiography discloses incomplete
or complete right-bundle-branch block in most patients or T-wave
inversions in leads V1-V3 (Fig. A). These conduction or repolarization
abnormalities are thought to be due to adipose infiltration
of the myocardium. Clinical diagnosis is based on detection
of predominantly right ventricular morphologic changes on imaging
studies. Echocardiography is an effective tool to demonstrate
the characteristic abnormal structure of the right ventricle,
including hypokinesis, massive dilatation, and a “parchment-thin”
wall (Fig. B). In addition, tricuspid regurgitation and paradoxic
ventricular septal wall motion are common. Pathologically, there
is variable infiltration or replacement of the right ventricular
myocardium by adipose and fibrous tissue.
The importance of right ventricular dysplasia is its association
with sudden death, with an incidence of up to 20 percent in
some series.
Therapy therefore is focused on the prevention of sudden death
with implantation of automatic internal cardioverter-defibrillators.
FIGURE
206
UhIs' anomaly. A. Twelve-lead electrocardiogram
demonstrating characteristic right bundle branch block with
T-wave inversions in leads V1-V3. B. Two-dimensional echocardiographic
four-chamber view demonstrating massive right ventricular dilation
"parchment-thin" wall
FIGURE
207
Gradient-echo image of a patient with
arrythmogenic RV dysplasia (ARVD). Note the focal region of
thinning of the right.
Ablation of Ventricular Tachycardia Foci
One of the most demanding of the catheter-ablative
techniques is attempted ablation of foci initiating ventricular
tachycardia. For this procedure, multipolar electrode catheters
are inserted into the right ventricle, coronary sinus, and left
ventricle. Ventricular tachycardia is induced by using standard
stimulation protocols, and the catheters are manipulated within
the ventricles to determine the earliest ventricular endocardial
electrogram (during ventricular tachycardia) in relation to
at least three reference orthogonal surface leads. Ventricular
overdrive pacing is used in an attempt to entrain the tachycardia
and to prove that the earliest endocardial potentials precede
(rather than follow) the tachycardia complex. In addition, the
putative focus of ventricular tachycardia is paced in an effort
to determine whether the paced complexes are identical or similar
to the induced tachycardia. The latter procedure is known as
pace mapping. For patients with ventricular tachycardia due
to coronary artery disease, concealed entrainment is manifest
by a prolonged paced spike to QRS, a paced QRS identical to
spontaneous tachycardia, and a post-pacing interval identical
to the spontaneous ventricular tachycardia cycle length, which
appears to best identify the critical slow zone for the ventricular
tachycardia reentrant circuit. Once the putative isthmus is
found, one or more radiofrequency applications are delivered
from the distal electrode near this endocardial site to a chest-wall
patch.
A subset of patients with ventricular tachycardia and structural
heart disease particularly amenable to catheter ablation are
those with bundle-branch reentrant arrhythmias. These patients
are recognized by having a left intraventricular conduction
delay or a frank pattern of left bundle-branch block. The majority
have an associated cardiomyopathy, and all have prolonged infranodal
conduction. In these patients, the tachycardic mechanism involves
bundle-to-bundle conduction. 27 Catheter cure may be achieved
by ablation of the right bundle branch. The right bundle usually
is draped superficially over the right septal surface, and the
right bundle potential usually is located easily. The right
bundle may be ablated either by direct current or preferably
by radiofrequency discharges. 27 Even after successful ablation
of the right bundle branch, further electrophysiologic testing
is in order to exclude ventricular tachycardia emanating from
myocardial sources.
Other forms of ventricular tachycardia that may be particularly
amenable to catheter ablation are those occurring in patients
without structural cardiac disease. These patients present with
tachycardia emanating from either the right ventricular outflow
tract~ or from the inferior left septum.29 Patients with tachycardia
emanating from the right ventricular outflow show a pattern
of left bundle-branch block with an inferior axis. The arrhythmia
is often exercise-induced and may respond to carotid massage
or treatment with adenosine or beta blockers. This arrhythmia
is thought to be a cyclic AMP-dependent triggered arrhythmia.
The hallmark of proper ablation includes detection of early
areas in the outflow tract and a precise correspondence between
the paced map and spontaneous ventricular tachycardia. Another
important site of ventricular tachycardia in normal hearts may
emanate from the left apical septum. This arrhythmia is characterized
by a pattern of right bundle-branch block associated with a
left superior axis. This arrhythmia most often responds to intravenous
verapamil. Ablative approaches include recording a Purkinje
potential just in front of the QRS complex and/or a paced map
that corresponds to the spontaneous tachycardia.
Ablation of ventricuiar tachycardia
Successful ablation was more frequent
in those with ventricular tachycardia associated with no structural
heart disease (85 percent), including those with right ventricular
outflow tract tachycardia or left septal tachycardia, compared
with ablation for ventricular tachycardia associated with coronary
artery disease (58 percent) or idiopathic cardiomyopathy (62
percent).Major complications included a post-procedural death
from presumed respiratory failure and cardiac tamponade, pulmonary
edema, systemic emboli, AV block, and femoral artery thrombosis.
ADVANTAGES AND DISADVANTAGES of CATHETER-ABLATIVE TECHNIQUES
Advantages
The use of catheter-ablative techniques has
greatly affected our approach to the management of patients
with supraventricular tachycardia. Catheter ablation of the
AV junction has replaced the need for surgical ablation of the
His bundle for patients with atrial arrhythmias refractory to
drug therapy. Furthermore, use of catheter procedures allows
cure of patients with reentrant supraventncular arrhythmias.
The initial reports suggest a cure rate of 90 to 100 percent
with minimal serious adverse effects. For selected patients
with ventricular tachycardia, catheter-ablative procedures may
obviate the need for surgical intervention. This is particularly
true for patients with bundle-branch reentry or for those with
right ventricular outflow tract or left septal tachycardias
Disadvantages
The chief disadvantage of AV junctional
ablation is the need for chronic cardiac pacing after successful
ablation. Another serious adverse effect is the reported 2 to
4 percent incidence of polymorphous ventricular tachycardia
occurring in the post-ablative period. This arrhythmia is more
common in patients with severe myocardial disease, bradycardia,
and electrolyte abnormalities, and may be prevented by temporarily
pacing the heart at relatively fast rates immediately after
ablation. The chief complication reported for patients undergoing
AV modification procedures for AV nodal reentry is the risk
of complete AV block. Attempted ablation of the slow AV nodal
pathway promises to diminish or obviate this risk.
The risks of catheter ablation of accessory pathways appears
to be related to the pathway site. Reported complications for
left free wall pathways include the risk of systemic embolization,
tamponade, or damage to the left circumflex coronary artery.
Ablation of septal pathways carries the risk of causing inadvertent
complete AV block. Fortunately, the risk of significant 12 complications
appears to be on the order of approximately 2 percent.
Major complications have been reported in the use of catheter-ablation
treatment of ventricular tachycardia. Such compli cations include
the risk of cerebrovascular accidents, damage to the aortic
valve, or tamponade.
L. Cocaine
M. Familial
Mutations in sarcomere protein genes (cardiac beta-myosin heavy chain and cardiac troponin T and actin) account for about 10 per cent of cases of familial dilated cardiomyopathy and are prevalent in families with early-onset ventricular dilatation and dysfunction resulting in congestive heart failure and sudden death in children and young adults.
Because distinct mutations in sarcomere proteins cause either dilated or hypertrophic cardiomyopathy, the effects of mutant sarcomere proteins on muscle mechanics must trigger two different series of events that remodel the heart
(see figure 126).
Reference:Kamisago,M.and Others,The New England Journal of Medicine,12/7/00,Vol.343,No.23,PP168-169
Genetic Causes of Cardiomyopathies in Humans and Animal Models
The ability to genetically manipulate
the cardiovascular system has made it possible to investigate
the role of a number of genes in the developing and adult mouse
heart (for a review, see Robbins). The discovery that mutations
in sarcomeric proteins lead to HCM has made it possible to generate
ani
mal models for this disease. In the case of myosin mutations,
a single genetic defect initiates a pathway that ultimately
leads to hypertrophy and then in males results in late decompensation
and ventricular dilatation. Multiple gene mutations have now
been associated causally with familial dilated cardiomyopathies.
A serendipitous genetic model of dilated cardiomyopathy and
heart failure (myf 5 mice) has been generated by activation
of a skeletal muscle genetic program in the heart. These mice
have a dilated cardiomyopathy phenotype characterized by progressive
myocardial dysfunction and dilatation. They develop the clinical
syndrome of heart failure, and they have an extraordinarily
high (>90 percent at 260 days) heart failure-related mortality
Another serendipitous genetic model of dilated cardiomyopathy
is the muscle LIM protein (MLP) knockout mouse. 2 MLP is a positive
regulator of muscle differentiation that is ordinarily expressed
at high levels in the heart and which may be involved in myofibrillar
protein assembly along the actin-based cytoskeleton MLP knockout
mice exhibit typical features of dilated cardiomyopathy, including
decreased systolic and diastolic function and /3-adrenergic
These characteristics make this model very useful in assessing
the mechanisms that lead to the development and progression
of myocardial failure. Thus, in transgenic mouse models, both
altered expression of contractile proteins and perturbation
of myocyte cytoarchitecture can lead to the dilated cardiomyopathy
phenotype.
There are several additional transgenic mouse models of cardiomyopathy
that may be more relevant to the production of a dilated phenotype
in humans. Three of them involve overexpression of components
of the adrenergic receptor pathway, the heterodimeric G-protein
a, subunit (Gas)23,z4 and the /31_21,2' and /32-adrenergic receptors.
These B-adrenergic pathway transgenic mouse models exhibit similar
histopathology consisting of myocyte hypertrophy and increased
fibrosis, evidence of apoptosis, systolic and diastolic dysfunction,
and ultimately, development of LV dilatation.
Several transgenic models of concentric or symmetrical LV hypertrophy
have now been reported, including overexpression of the protooncogenes
ras and myc, a,-adrenergic receptors, the heterodimeric G-protein
a subunit (G-alpha-4),and protein kinase C (PKC) The mechanisms
for the induction of increased ventricular wall thickness are
diverse, inasmuch as the ras, a,receptor, G-alpha, and PKC overexpressors
exhibit true cellular hypertrophy with an increase in cell size
, whereas the myc animal exhibits cardiac myocyte hyperplasia
The HCM phenotypes discussed earlier illustrate the principle
that apparently diverse signals can culminate in the same phenotype,
presumably by converging on final common pathways.
Multiple gene defects have been identified that can produce
a dilated cardiomyopathy in humans, as discussed in more detail
in the section on familial forms of dilated cardiomyopathy.
As listed in Table
66-2, these include mutations in the cardiac a actin, 34
desmin, 35 dystrophin, 36, 31 and lamin3s.39 genes.
Polymorphic Variation in Modifier Genes
Genes exhibit polymorphic variation; i.e., normal variants of
genes exist in the population that are of slightly different
size or sequence. Some gene polymorphisms are associated with
differences in function of the expressed protein gene product,
and some of these differences in function likely account for
"biologic variation" routinely encountered in population
Examples of "modifier" genes that may have an impact
on the natural history of a dilated cardiomyopathy (see Table
66-2) include the angiotensin-converting enzyme (ACE) DD
genotype, where individuals are homozygous for the "deletion"
variant, which is associated with increased circulating"
and cardiac tissue41 ACE activity. The DD genotype appears to
increase the extent of hypertrophy in HCM and may be a risk
factor for early remodeling after MI and for the development
Other potentially important polymorphic variants that may influence the natural history of a cardiomyopathy involve the angiotensin AT-1 receptor and B2-adrenergic receptors.
Altered, Maladaptive Expression of a Completely Normal Gene
The third way in which altered gene expression
can contribute to the development of a cardiomyopathy is altered,
maladaptive expression of a completely normal "wild type"
gene." This occurs most commonly in the context of progression
of heart muscle disease and myocardial failure, which is the
natural history of virtually all cardiomyopathies once they
are established. Examples in this category include downregulation
of B1-adrenergic receptors, alpha-myosin heavy chain (alpha-MHC),
and sarcoplasmic reticulum Ca2+ ATPase genes and upregulation
in the atrial natriuretic peptide (ANP),myosin heavy chain (B-MHC),
ACE, tumor necrosis factor (TNF-alpha), endothelin, B-adrenergic
receptor kinase (B-ARK) genes. These concepts are discussed
PATHOPHYSIOLOGIC PROCESSES INVOLVED IN MYOCARDIAL DYSFUNCTION,
REMODELING, AND THEIR PROGRESSION
Tissue preparations and myocytes isolated from failing human
hearts exhibit evidence of decreased contractile function Assuming
that loading conditions and ischemia are not adversely affecting
cardiac myocyte function, in the setting of chronic systolic
dysfunction from a dilated cardiomyopathy, progressive myocardial
failure is most likely caused by myocardial cell loss or changes
in the gene expression of proteins that regulate or produce
muscle contraction. Figures 126-b and 126-c summarize these
general points and emphasize the central roles of the renin-angiotensin
system (RAS) and the adrenergic nervous system (ANS) in promoting
cell loss, growth and remodeling, and altered gene expression.
Myocardial Dysfunction and Remodeling
due to Altered Expression of Contractility Regulating Genes
and Changes in Sarcomeric Assembly
Gene expression can be defined, broadly, as the expression of
a fully or normally functioning protein gene product or, more
narrowly (and commonly), as the steady-state abundance of a
gene's mRNA transcript. Using either definition, numerous
FIGURE
126-b Relationship of neurohormonal
activation and production of cardiac myocyte loss due to apoptosis
and necrosis and altered gene expression. Cell loss and altered
gene expression result in more myocardial dysfunction, and a
vicious cycle is established. RAS = renin angiotensin system;
ANS = adrenergic nervous system.
FIGURE 126-c Heart failure compensatory mechanisms that are activated to support the failing heart. Light-colored areas indicate physiologic mechanisms that stabilize pump function.
abnormalities of gene expression of normal, wild-type genes
have been demonstrated in the failing human heart, as discussed
earlier. In order to characterize the abnormalities that may
account for progressive myocardial dysfunction and remodeling,
it is useful to subdivide them into two general categories,as
shown in table 66-3 below.
The first category encompasses mechanisms that subserve intrinsic
function, or the mechanisms responsible for contraction and
relaxation of the heart in the basal or resting state. Intrinsic
function is defined as myocardial contraction and relaxation
in the absence of extrinsic influences, such as neurotransmitters
or hormones.
The second general category is modulated function, which comprises
the mechanisms responsible for the remarkable ability of the
heart to increase or decrease its performance dramatically (by
2- to 10-fold) and rapidly in response to various physiologic
or physical stimuli. Other critical organs such as the brain,
kidney, and liver do not exhibit this quality. Modulated function
is defined as stimulation or inhibition of myocardial contraction
or relaxation by endogenous bioactive compounds, including neurotransmitters,
cytokines, autocrine/paracrine substances, and hormones.
In the failing human heart, changes are present in the expression
of genes potentially responsible for both general types of myocardial
function . Abnormalities of intrinsic function include the factors
responsible for an altered length-tension relation a blunted
force-frequency response and/or the signals responsible for
abnormal cellular and chamber remodeling . In the case of the
abnormal forcefrequency and length-tension responses, the evidence
favors abnormal contractile function of individual cardiac myocytes.As
shown in table 66-3,these abnormalities likely reside in the
contractile proteins or their regulatory elements, mechanisms
involved in excitation-contraction coupling, or the cytoskeleton.
However, within these possibilities for altered intrinsic function,
there is not currently a consensus as to which specific abnormalities
are present in idiopathic dilated cardiomyopathy (IDC), the
most common form of heart failure studied in humans. For cellular
remodeling, in both human ventri- cles and animal models, the
assembly of sarcomeres in series leads to a myocyte that is
markedly increased in length but not in diameter, which contributes
to remodeling at the chamber level. Such remodeling places the
chamber and the myocyte at an energetic disadvantage because
of the attendant increase in wall stress, which is one of the
major determinants of myocardial oxygen consumption. Inadequate
myocyte energy production, particularly associated with key
subcellular ion flux mechanisms or the myosin ATPase cycle,
in turn would contribute to myocyte contractile dysfunction.
Moreover, the hypertrophy process itself leads to a qualitative
change in contractile protein gene expression (induction of
a "fetal" gene program) that reduces contractile function
. On the other hand, cardiac myocyte contractile dysfunction
likely plays a role in the remodeling process, inasmuch as medical
treatment that improves intrinsic myocardial function can reverse
remodeling.' Thus contractile dysfunction and remodeling at
the cellular level are intimately related to the progressive
contractile dysfunction and chamber enlargement that define
the natural history of myocardial failure." These concepts
are summarized in Fig.
126-d.
In contrast to abnormalities of intrinsic
function, a consensus has been reached on several specific abnormalities
in the stimulation component of modulated function. Most of
these changes concern B-adrenergic signal transduction. The
ability of beta adrenergic stimulation to increase heart rate
and contractility is markedly attenuated in the failing heart
due to multiple changes at the level of receptors, G-proteins,
and adenylyl cyclase. This produces a major abnormality in the
stimulation component of modulated function. In addition, the
inhibition component of modulated function is also abnormal
in the failing heart, due to a reduction in parasympathetic
drive.
There is obviously overlap between the two major subdivisions
of myocardial function. Recent data indicate that even in the
absence of adrenergic stimulation, beta-adrenergic receptors
have intrinsic activity That is, a small number of receptors
are in an activated state without agonist occupancy and as such
can support intrinsic myocardial function ....ft. Thus overexpression
of human B2-adrenergic receptors is able to markedly increase
intrinsic myocardial function, as is enhancement of sarcoplasmic
reticulum calcium uptake and release by genetic ablation of
the phospholamban gene. The recent realization that active state,
agonist-unoccupied beta-adrenergic receptors can modulate intrinsic
myocardial function is the reason why the "R-G-adenylyl
cyclase" mechanism appears in both categories in Table
ProgressiveMyocardial Dysfunction and Remodeling due to Loss of Cardiac Myocytes
The second general mechanism by which myocardial function may be adversely affected is by loss of cardiac myocytes,, which also my play a role in the progresssion of ventricular dysfunction in dilated cardiomyopathies. Cardiac myocyte loss can occur via toxic mechanisms producing necrosis or by "programmed cell death" producing apoptosis. Apoptosis, which is likely due to a combination of growth signaling and cell cycle dysregulation, has been described in end-stage IDC, as well as in the B-1,-adrenergic receptor, the G alpha- s overexpressor transgenic mice, and in models of hypertrophy. However, the human hearts with IDC or ischemic cardiomyopathy were taken from very late stage, literally dying patients maintained on multiple powerful intravenous inotropic medications,and it is not clear if apoptosis plays a significant role in remodeling and/or chamber systolic dysfunction
IMPORTANCE OF "COMPENSATORY"
As depicted in Fig.
126-b and Fig.
126-c, there is now a large body of information supporting
the idea that activation of the ANS and RAS compensatory mechanisms
contributes to, or is responsible for, the progressive nature
of both myocardial failure and the natural history of the heart
failure clinical syndrome. This evidence includes the observations
that activation of both these systems is associated with progression
of myocardial dysfunction and the heart failure syndrome and
clinical trial data that consistently demonstrate that inhibition
of these systems can prevent deterioration in or improve myocardial
function as well as reduce mortality . Despite the fact that
in human heart failure we now know that chronic
| TABLE 66-3 General Categorization
of Myocardial Function Intrinsic (Function in the Absence of Neural or Hormonal Influence) |
| • Contractile proteins • E-C coupling mechanisms • R-G-adenylyl cyclase pathways • Bioenergetics • Cytoskeleton • Sarcomere and cell remodeling |
ABBREVIATIONS: E-C = excitation-contraction;
R-G = receptor-G-protein. Modulated (Function that May Be Stimulated or Inhibited by Extrinsic Factors Including Neurotransmitters, Cytokines, or Hormone) • R-G-adenylyl cyclase
pathways |
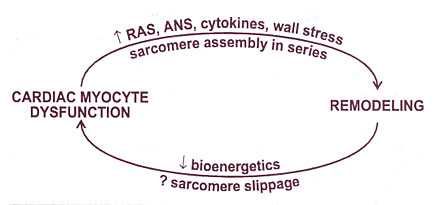
Fig.126-d: Relationship between progressive myocardial dysfunction and remodeling.RAS= renin angiotensin system;ANS =adrenergic nervous system.
activation of the ANS and RAS contributes to the progressive nature of myocardial dysfunction, we know virtually nothing about how these systems adversely affect the biology of the cardiac myocyte. What we do know is that mechanisms within both general categories outlined in Table 66-3 below (fig. 126-d) must be involved in the adverse myocardial effects mediated by the ANS and RAS. This is so because modulated function may be improved by treatment with ACE inhibitors or beta-blocking agents. Progressive myocardial dysfunction and remodeling are attenuated by both beta-blocking agents and ACE inhibitors, and in cardiomyopathies, intrinsic myocardial function is improved and remodeling is reversed by chronic treatment with beta-blocking agents. Additionally, mortality in chronic heart failure is directly related to activation of the ANS and RAS and may be related to activation of other neurohormonal or autocrine/paracrine systems as well.
Regardless of the type or cause of dilated cardiomyopathy, an
initial myocardial insult resulting in this phenotype exhibits
common pathophysiologic features that are summarized in Fig.
126-c. That is, a myocardial insult that produces systolic
dysfunction will be followed by the initiation of processes
designed to temporarily stabilize pump function. The possible
mechanisms available for such stabilization are in fact limited.
As shown in Fig.
126-b, in chronological order of their action, they are
an increase in heart rate and contractility mediated by an increase
in cardiac beta-adrenergic signaling (produced within seconds
of the onset of pump dysfunction), volume expansion in order
to use the Frank-Starling mechanism to increase stroke volume
(evident within hours of the onset of pump dysfunction), and
cardiac myocyte hypertrophy to increase the number of contractile
elements (evident within days to weeks of the onset of pump
dysfunction). As shown in Fig.
126-b, these compensatory adjustments are largely accomplished
by activation of the RAS and ANS. However, despite the short-term
(days to months) stability achieved via these mechanisms, they
ultimately prove harmful. The best evidence that chronic, continued
activation of the RAS and ANS contributes to progressive myocardial
dysfunction and remodeling comes from clinical trials where
both inhibitors of the RAS (ACE inhibitors) and ANS (beta adrenergic
receptor-blocking agents) prevent these two phenomena, and beta-blocking
agents actually may reverse remodeling and progressive systolic
dysfunction, as alluded to.
Much current work is focused on the precise pathophysiologic
mechanisms by which activation of the RAS and ANS produces remodeling
and adverse effects on myocardial function. Some of the possibilities
are given in Fig.
126-b, and they include an exacerbation of ischemia and/or
energy depletion leading to cell loss via necrosis, cell loss
by programmed cell death, direct promotion of hypertrophy and
remodeling through stimulation of cell growth, and alterations
in cardiac myocyte gene expression. A key feature of the schema
shown in Fig.
126-b is the process of remodeling. Virtually all dilated
cardiomyopathies undergo this process, which is characterized
by progressive dilatation, progressive myocardial systolic dysfunction
in viable segments, and a chamber shape change whereby the ventricle
becomes less elliptical and more round 6°63 As shown in Fig.
126-d, this places the ventricle at an energetic disadvantage,
which likely contributes to further myocardial dysfunction,
which then contributes to progressive remodeling. The latter
observation is based on data obtained with beta-adrenergic blocking
agents, which produce an improvement in systolic dysfunction
that can be detected prior to a reversal in remodeling .As emphasized
by Fig.
126-d, each myocardial degenerative process likely begets
the other, leading to an inexorably progressive deterioration
in myocardial performance and clinical condition.
SCOPE OF DILATED CARDIOMYOPATHIES
The number of cardiac or systemic processes
that can produce or are associated with a dilated cardiomyopathy
are plentiful and remarkably varied, as shown in Table 66-4.
The dilated phenotype is by far the most common form of cardiomyopathy,
comprising over 90 percent of subjects referred to specialized
centers. In the United States, the most common dilated cardiomyopathy
is ischemic dilated cardiomyopathy, or the cardiomyopathy that
follows MI. Other common secondary dilated cardiomyopathies
are hypertensive and valvular dilated cardiomyopathies, both
produced in part by chronically increased wall stress. The primary
cardiomyopathy, IDC, is another relatively common dilated phenotype,
as discussed below.
SELECTED, COMMON TYPES OF DILATED CARDIOMYOPATHIES
Ischemic Cardiomyopathy
DEFINITION/DIAGNOSIS
Ischemic cardiomyopathy is defined as
a dilated cardiomyopathy in a subject with a history of MI or
evidence of clinically significant (i.e., approximately70 percent
narrowing of a major epicardial artery) coronary artery disease,
in whom the degree of myocardial dysfunction and ventricular
dilatation is not explained solely by the extent of previous
infarction or the degree of ongoing ischemic. In other words,
an ischemic dilated cardiomyopathy is present when a post-MI
left ventricle experiences remodeling and a drop in ejection
fraction.
DISTINCT PATHOPHYSIOLOGY
Dilatation of the left ventricle and a decrease in ejection
fraction occurs in 15 to 40 percent of subjects within 12 to
24 months following an anterior MI and in a smaller percentage
of subjects following an inferior MI. Based on limited data
'41 it is tempting to speculate that the subjects who undergo
the remodeling process and develop an ischemic dilated cardiomyopathy
are individuals with particularly heightened compensatory mechanisms
(see Fig.
126-b and Fig. 126-c), perhaps as a result in polymorphic variation
in these systems. As discussed earlier, the remodeling process
is an attempt by the compromised ventricle to increase its performance
by increasing stroke volume, but ultimately, it correlates with
an adverse outcome in the long term.
The gross pathology of ischemic cardiomyopathy includes transmural
or subendocardial scarring, representing old MIs, that may comprise
up to 50 percent of the LV chamber. The histopathology of the
noninfarcted regions is similar to changes that occur in IDC,
as discussed below.
PROGNOSIS
Several studies have concluded that ischemic
cardiomyopathy patients have a worse prognosis than subjects
with a "nonischemic" dilated cardiomyopathy, probably
because the risk of ischemic events is added to the risk of
TREATMENT
The treatment of ischemic dilated
cardiomyopathy and chronic heart failure is covered in detail
in elsewhere1. In general, treatment consists of the use of
ACE inhibitors in asymptomatic or symptomatic patients, the
use of diuretics in volume-overloaded subjects, and the use
of digoxin in subjects who remain symptomatic on the former
medications. An emerging treatment strategy is the use of beta-adrenergic
blocking agents in mild to moderately symptomatic subjects ,
whereas in both ischemic and nonischemic dilated cardiomyopathies,
second- and third-generation compounds improve LV function,
reduce hospitalizations, and lower mortality. Additionally,
adjunctive therapy includes anticoagulation in subjects with
lower LV ejection fractions to prevent thromboembolic complications,
amiodarone to treat symptomatic arrhythmias, maintaining potassium
levels in the high normal (4.3-5.0 meq/L) range to prevent sudden
death, frequent clinic visits to adjust medications, and an
aggressive approach to treating ischemia, including revascularization.
Hypertensive Cardiomyopathy
DEFINITION/DIAGNOSIS
A hypertensive dilated cardiomyopathy
is diagnosed when myocardial systolic function is depressed
out of proportion to the increase in wall stress. In other words,
a subject presenting in heart failure with a hypertensive crisis
would not carry this diagnosis unless ventricular dilatation
and depressed systolic function remained after correction of
the hypertension. In addition to producing a "pure"
form of hypertensive cardiomyopathy, hypertension is a major
risk factor for heart failure from any cause.Within
the WHO/ISFC classification, "hypertensive heart disease"
may present in the "dilated'' ,"restrictive'', or
"unclassified" categories.
DISTINCT PATHOPHYSIOLOGY
The most important pathophysiologic element in hypertension in dilated cardiomyopathy is sustained increased systolic wall stress. Interestingly, in both systolic pressure overloaded right and left ventricles, phenotypic expression is qualitatively variable and can include dilatation and systolic dysfunction without increased wall thickness, increased wall thickness, concentric hypertrophy with or without systolic dysfunction, and systolic dysfunction without concentric hypertrophy. Other contributors to the pathophysiology of hypertensive cardiomyopathies are local neurohormonal mechanisms.
PROGNOSIS
The prognosis depends on the presence of other comorbid conditions such as diabetes mellitus and coronary artery disease, as well as the extent of control of afterload. Compared with other forms of cardiomyopathy, in the absence of comorbid conditions, the prognosis of hypertensive cardiomyopathy in subjects whose afterload is controlled is probably better than for most other types of dilated cardiomyopathy.
TREATMENT
The treatment is as for ischemic dilated cardiomyopathy, except that afterload must be vigorously controlled. This consists of the addition of pure antihypertensive vasodilators such as amlodipine or a-blocking agents to standard heart failure therapy.
Valvular Cardiomyopathy
DEFINITION/DIAGNOSIS
A valvular cardiomyopathy occurs when a valvular abnormality is present and myocardial systolic function is depressed out of proportion to the increase in wall stress. This most commonly occurs with left-sided regurgitant lesions (mitral regurgitation and aortic regurgitation), less commonly occurs with aortic stenosis, and never occurs as a consequence of pure mitral stenosis.
DISTINCT PATHOPYSIOLOGY
The classic explanation for the typical phenotypes observed in valvular cardiomyopathies relates to exposure to different types of wall stress."' Within this construct, the pattern of eccentric hypertrophy derives from increased diastolic wall stress.Thus long-standing mitral regurgitation most commonly results in compensated eccentric hypertrophy that can progress to a dilated failing phenotype. Aortic regurgitation is a particularly poorly tolerated hemodynamic insult because wall stress is increased in both systole and diastole, and when decompensation occurs, ventricular volume will be increased with or without increased wall thickness. Aortic stenosis classically results in compensated concentric hypertrophy, but when decompensation occurs, a variety of phenotypes can be observed that are similar to hypertensive cardiomyopathies. A disturbing and fairly commonly observed phenomenon is the development of a dilated cardiomyopathy after surgical correction of mitral and sometimes aortic valve disease in subjects who preoperatively had only mild LV dysfunction. These cases are likely due to the superimposition of myocardial damage resulting from open heart surgery and/or underlying dysfunction
PROGNOSIS
The prognosis is variable and depends on the number of associated conditions, the nature and extent of the valvular abnormality, and most important, the severity of the cardiomyopathy at the time of surgical correction (see below). In general, severely depressed myocardial function will not improve much with surgical repair of aortic regurgitation or mitral regurgitation, but the prognosis is likely to be improved because of elimination of some of the hemodynamic insult. Replacement of the mitral valve should not be attempted in the majority of subjects with severe mitral regurgitation and LV ejection fractions less than 25 percent because of prohibitively high operative/perioperative mortality rates. On the other hand, there is no impairment of LV systolic function severe enough to preclude valve replacement of severe aortic stenosis, since function invariably will improve on relief of the hemodynamic insult, and the prognosis is relatively good.
TREATMENT
The treatment of a valvular dilated cardiomyopathy is surgical valve replacement or repair as soon as the cardiomyopathy is detected. Catheter valvuloplasty may be an option for severe aortic stenosis (AS) patients who are not good surgical candidates for reasons other than heart failure. Medical treatment may be the only option in subjects with aortic insufficiency or mitral regurgitation whose LV function is severely impaired. The medical treatment of either disorder should be as mentioned earlier for ischemic cardiomyopathy plus aggressive afterload reduction, usually hydralazine/nitrates on top of ACE inhibitors. The calcium channel blocker amlodipine is another option for afterload reduction, particularly for aortic insufficiency, where calcium channel blocker therapy has been shown to improve survival.
Idiopathic Dilated Cardiomyopathy, Including Familial Forms
DEFINITION/DIAGNOSIS
IDC is diagnosed by excluding significant coronary artery disease,
valvular abnormalities, and other causes. IDC is a relatively
common cause of heart failure, with an estimated prevalence
rate of 0.04 percent and incidence rates varying from 0.005
to 0.006 percent . The true incidence of IDC is undoubtedly
higher, owing to the fact that subjects may remain asymptomatic
until marked ventricular dysfunction has occurred. The incidence
of IDC increases with age, and males are afflicted at a higher
rate than are females. As discussed below, histologic features
are nonspecific and consist of myocardial cell hypertrophy and
varying amounts of increased interstitial fibrosis. Although
the diagnosis is not difficult, problems arise when an apparent
IDC presents in someone with a history of hypertension or excessive
alcohol intake. In such cases, it is best to reassign the etiology
to alcohol only when the intake has exceeded 80 g/day for males
and 40 g/day for females for more than 5 years and to hypertensive
heart disease when blood pressure has been uncontrolled and
high (>160/100 mmHg), as well as sustained (for years). All
subjects with an unexplained dilated cardiomyopathy need a thyroid-stimulating
hormone (TSH) determination done to exclude hypo- or hyperthyroidism,
and subjects with diastolic dysfunction need to have an infiltrative
process excluded. As discussed below, this is best done by performing
DISTINCT PATHOPHYSIOLOGY
IDC may be familial in as many as 35 to 50 percent of the patients
when first-degree relatives are carefully screened.' The analysis
of the phenotype identifies a wide range of clinical and pathologic
forms indicating genetic heterogeneity. Accordingly, several
chromosomal assignments for gene location have been made, and
recently, as shown in Table
66-2, several genes have been identified . The majority
of familial patients present with autosomal dominant inheritance
and a phenotype characterized by low and age-related penetrance
(which is the proportion of carriers who manifest the disease).
It is estimated that only 20 percent of gene carriers under
the age of 20 display the disease phenotype. Autosomal dominant
dilated cardiomyopathy can be due to mutations of the cardiac
actin or desmin gene, but in the majority of cases the disease
gene is still unknown. The detection of an altered creatine
kinase level can indicate the existence of a subclinical skeletal
muscle disease. In these patients, an X-linked inheritance suggests
mutations in the dystrophin gene whereas an autosomal dominant
transmission and the presence of conduction defects and arrhythmia
suggests mutations in the lamin A/C gene . In laminopathy, the
phenotype of the affected relatives can be very variable, from
a pure IDC to a mild Emery-Dreifuss-like or limb-girdle-like
muscle dystrophy . Skeletal muscle and endomyocardial biopsy
are diagnostic in X-linked dilated cardiomyopathy, showing abnormalities
of dystrophin protein expression by immunocytochemistry. Finally,
autosomal recessive transmission of dilated cardiomyopathy occurs
in mutations of sarcoglycan genes, which encode for dystrophin
complex-associated proteins.
Dystrophin, sarcoglycans, desmin, and lamin are cytoskeletal
proteins. The contractile protein cardiac a-actin also has a
forcetransmission or cytoskeletal role. Other data support the
hypothesis that IDC could represent, in the majority of cases,
a disease of the cytoskeleton; absence of the protein metavinculin
in the myocardium was reported in one IDC patient, and as discussed
earlier, a dilated cardiomyopathy can be created in mice22 or
is present in a hamster line related to mutations in cytoskeletal
genes. However, as discussed earlier, it appears that other
genetic abnormalities such as mutations in contractile proteins
and overexpression of beta-adrenergic receptors or Gas24 also
can produce a dilated phenotype.
In children, X-linked familial IDC suggests mutation in the
G4.5 or tafazzin gene, particularly if associated with certain
other signs (such as endocardial fibroelastosis, neutropenia,
short stature, or skeletal muscle abnormalities). The function
of tafazzin is still unknown. In mitochondrial DNA (mtDNA) mutations,
myocardial dysfunction usually is associated with multiorgan
involvement (encephalopathy, lactic acidosis, skeletal muscle
abnormalities, retinitis pigmentosa, etc.). It is still unclear
whether a mtDNA mutation can lead to an isolated IDC phenotype
in adults.
Although still incomplete, new knowledge on the genetics of
IDC has important clinical implications. The frequency of familial
forms indicates the need of family screening in IDC, which can
allow genetic counseling, an early detection of the disease,
and early therapeutic interventions in affected relatives. The
complexity of the phenotype requires an accurate skeletal muscle
investigation, which can direct the diagnosis toward a specific
type of familial myopathy. Finally, family investigations require
more sensitive diagnostic criteria 131 that are able to detect
minor cardiac abnormalities as initial signs of the disease.
These include initial dilatation without marked systolic dysfunction,
arrhythmia, and isolated wall and other abnormalities.
The major morphologic feature of IDC on postmortem examination
is dilatation of the cardiac chambers . One ventricle (usually
the left) may be more dilated than the other ventricle. The
weight of the heart is increased in IDC, with a mean cardiac
weight of 551 g for women and 632 g for men. Although there
is an increase in muscle mass and myocyte cell volume in IDC,
LV wall thickness is usually not increased because of the marked
dilatation of the ventricular cavities. Grossly visible scars
may be present in either ventricle, and while most scars are
small, some may be large and transmural. Scarring occurs in
the absence of significant narrowing of the epicardial coronary
arteries. In most cases, the degree of fibrosis does not appear
to be extensive enough to cause changes in systolic or diastolic
function. Intracardiac thrombi and mural endocardial plaques
(from the organization of thrombi) are present at necropsy in
more than 50 percent of patients with IDC. The effect of anticoagulation
on the incidence of thrombi has not been studied carefully,
but systemic and pulmonary emboli are more frequent in patients
with ventricular thrombi or plaques.
The characteristic findings of IDC on microscopy are marked
myocyte hypertrophy, very large, bizarrely shaped nuclei (Fig.
126-e) , increased interstitial fibrosis (see Fig.
126-e), myocyte atrophy, and myofilament loss. In isolated
cardiac myocytes, the major cellular phenotypic change is marked
increase in cell length without a concomitant increase in diameter.
As described earlier, this cellular lengthening or remodeling
contributes to the chamber remodeling/dilatation that characterizes
IDC and other cardiomyopathies. These morphologic changes in
IDC are not specific and are generally found in secondary cardiomyopathies
such as in the noninfarcted regions of ischemic dilated cardiomyopathy.
Also, the morphometric changes in IDC do not correlate with
the severity of illness. Ultrastructural abnormalities such
as mitochondrial changes, T-tubular dilatation, and intracellular
lipid droplets may be observed in IDC but also can be observed
in other forms of heart disease . There may be interstitial
parenchymal and perivascular focal infiltrates of small lymphocytes.
The lymphocytic infiltrates that are present on histologic examination
in IDC are not associated with adjacent myocyte damage, in contrast
to myocarditis where adjacent myocyte necrosis is observed.
Fibrosis is nearly always present in IDC, and its pattern is
quite variable from a fine perimyocytic distribution to coarse
scars indistinguishable from those present in chronic ischemia.
However, small intramural arteries and capillaries are structurally
normal in IDC.
A number of immune regulatory abnormalities have been identified
in IDC, including humoral and cellular autoimmune reactivity
against myocytes, decreased natural killer cell activity, and
abnormal suppressor cell activity.These abnormalities suggest
that immune defects may be important etiologic factors in the
development of IDC. These findings, however, are not universally
present in patients with IDC, and some abnormalities are also
present in other types of heart muscle disease. For example,
an increase in the cardioselective M7 antimitochondrial antibodies
is found in both IDC and hypertrophic cardiomyopathy but not
in heart failure from coronary artery disease. The incidence
of some autoreactive antibodies, such as antinuclear and antifibrillary
antibodies, increases with the severity of heart failure. It
is likely that many of the antibodies detected in IDC and other
myocardial diseases do not have pathogenic relevance, but rather
are secondary to the primary degenerative process. However,
it is possible that certain antibodies present in IDC may have
important functional implications. For example, anti-beta1-adrenergic
receptor antibodies could modify beta-adrenergic receptor activity
and produce chronic increases in signal transduction that are
harmful to the failing heart. Disturbed energy metabolism from
antibodies to the ADP/ATP carrier of the inner mitochondrial
membrane is another potential pathogenetic autoimmune mechanism
; these antibodies are present in some individuals with IDC
and have been shown to impair metabolism and myocardial function.
There has been great interest in histocompatibility locusantigens
(HLAs) in IDC because these antigens are knownto be associated
with immune regulatory functions, and manyautoimmune diseases
are found to have positive HLA antigenicassociations. HLA associations
also have been identified in IDC;the frequency of HLA-B27, HLA-A2,
HLA-DR4, and HLADQ4 is increased compared with controls, and
the frequencyof HLA-DRw6 is decreased compared with controls.
Geneticabnormalities in the HLA region potentially could alter
immuneresponse and thereby increase disease susceptibility to
infectious agents such as enteroviruses. Thus the association
in IDCwith specific HLAs suggest a possible immunologic etiology
for thisdisease. However, these specificHLAs are present in
less than 50percent of patients with IDC, andthe heterogeneity
of these antigens does not point to a uniquesite for a putative
disease-associated gene. Thus, while the autoimmune hypothesis
is an attractivecandidate for the etiology of somecases of IDC,
it remains unproved.
A clinical and pathologic syndrome that is similar to IDC may
develop after resolution of viral myocarditis in animal models
and biopsy-proven myocarditis in human subjects. This has led
to speculation that IDC may develop in some individuals as a
result of subclinical viral myocarditis. Theoretically. an episode
of myocarditis could initiate a number of autoimmune reactions
that injure the myocardium and ultimately result in the development
of IDC. The
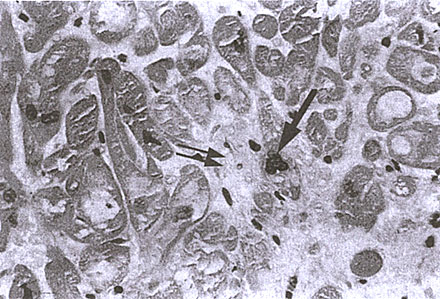
FIGURE 126-e: Right ventricular endomyocardial biopsy from a
subject with IDC. Note the increased nuclear size (arrow) and
the increased interstitial fibrosis.
abnormalities in immune regulation and the variety of antimyocardial antibodies present in IDC are consistent with this hypothesis. However, it is generally not possible to isolate an infectious virus or to demonstrate the presence of viral antigens in the myocardium of patients with IDC.'S4 Enteroviral RNA sequences are found in heart biopsy samples in IDC, but only in approximately one-third of patients. Furthermore, active myocardial inflammation is usually not detected in IDC. However, in controlled trials, corticosteroid therapy of patients with IDC does not result in significant clinical improvements . Importantly, recent experimental data have shown in vitro and in vivo that the enteroviral protease 2A is able to cleave dystrophin and disrupt the cytoskeleton in cardiac myocytes, providing a potential link between viral infection and a genetic model of the disease. Furthermore, analysis of human viruses other than enteroviruses suggests that adenoviruses, herpesvirus, and cytomegalovirus also can cause myocarditis and potentially IDC, particularly in children and young subjects. Further investigation will be necessary to understand the significance of these findings, particularly in the adult population.
Endomyocardial biopsy of the right or left ventricle may be a valuable diagnostic adjunct for diagnosing specific myocardial processes that can produce a dilated phenotype, such as myocarditis and infiltrative cardiomyopathies. Since several of these other dilated cardiomyopathies may have specific treatments and/or a different prognosis than IDC, endomyocardial biopsy may be warranted in many individuals presenting with a dilated cardiomyopathy. In the future, biopsy may be used more frequently to identify genetic disorders resulting in abnormal gene or protein expression, such as now can be done to diagnose Becker-Duchenne cardiomyopathy. Since special staining, electron microscopy, or molecular analysis of the biopsy material may be necessary, endomyocardial biopsy is best performed in specialized cardiomyopathy/heart failure centers.
PROGNOSIS
Several studies of the natural history of IDC have been conducted
. The prognosis is generally better than for ischemic cardiomyopathy,
and prior to the routine use of ACE inhibitors, survival was
approximately 50 percent in 5 years. The prognosis has been
improved substantially since then, inasmuch as ACE inhibition,
cardiac transplantation and beta-adrenergic blockade are all
effective treatments in this cardiomyopathy.
TREATMENT
The treatment of IDC is similar to that discussed earlier for
ischemic cardiomyopathy, except that there is no issue of revascularization.
The risk of thromboembolic complications may be higher than
in ischemic cardiomyopathy, resulting in a lower threshhold
for anticoagulation. Beta-Adrenergic blockade produces a quantitatively
greater degree of improvement in LV function compared with ischemic
cardiomyopathy either because there is a greater degree of adrenergic
activation or there is more viable myocardium to work with in
IDC. Approximately 10 percent of IDC subjects treated with beta-adrenergic
blockade will normalize their myocardial function, and this
form of treatment should be offered to all IDC patients who
do not have a contraindication before considering cardiac transplantation.
SELECTED SPECIFIC DILATED CARDIOMYOPATHIES
WITH UNIQUE MANAGEMENT ISSUE
Anthracydine Cardiomyopathy
DEFINITION/DIAGNOSIS
The commonly used and highly efficacious anthracycline antibiotic
anticancer agents doxorubicin and daunorubicin produce a dose-related
cardiomyopathy that may limit their clinical application. Within
the WHO/ISFC classification, an anthracycline cardiomyopathy
would most likely be in the "dilated" category, but
because the extent of dilatation initially may be minimal (see
below), it also could be in the "unclassified" category.
The cardiomyopathy produced by these agents depends on the total
cumulative dose, and for the more widely used compound doxorubicin
(Adriamycin), the incidence of heart failure due to cardiomyopathy
dramatically increases above total cumulative doses of 450 mg/m2
in subjects without underlying cardiac problems or other risk
factors.Prior mediastinal radiation involving the heart is a
powerful risk factor for anthracycline cardiomyopathy, and the
risk is also evident i f radiation treatment follows chemotherapy.
In subjects with risk factors, anthracycline cardiomyopathy
Although the diagnosis of anthracycline cardiomyopathy can be
made clinically, the definitive diagnosis depends on the demonstration
of a substantial number of cardiac myocytes exhibiting the characteristic
anthracycline effect. Tissue sampling is best done by endomyocardial
biopsy, which allows for "thin section" electron microscopic
processing of the sample and more definitive resolution of the
anthracycline effect with
light microscopy.
DISTINCT PATHOPHYSIOLOGY
In the absence of a tissue diagnosis,
anthracycline cardiomyopathy may be diagnosed clinically by
exclusion of other causes of cardiomyopathy in a subject who
has had at least 350 mg/m2 of doxorubicin or the equivalent
amount of another anthracycline. As shown in Fig.
126-f , the anthracycline cardiac myocytic lesion consists
of cell vacuolization progressing to cell dropout, and when
16 to 25 percent of the total number of sampled cells exhibit
this morphology, myocardial dysfunction results.
There are some distinguishing clinical features of anthracycline
cardiomyopathy that may relate to its pathophysiology. These
include a relative absence of hypertrophy and dilatation and
a higher heart rate (110-130 beats per minute) than is usually
encountered in ambulatory heart failure. The reasons for these
features are that the onset of symptoms may be relatively acute
(remodeling takes time to develop), and the anthracycline inhibits
contractile protein synthesis, reducing the amount of compensatory
dilatation and remodeling. In this situation, the only option
available for stabilizing cardiac output is increasing the heart
rate, since increasing stroke volume via a larger end-diastolic
volume has been precluded. The increased heart rate is produced
by a greater than expected hyperadrener-gic state,and so these
subjects may be exceptionally dependent on adrenergic support.
FIGURE 126-f Cardiac myocyte vacuolization in cases of Adriamycin cardiomyopathy classified on endomyocardial biopsy as grade 3 by the Billingham classification.
PROGNOSIS
The prognosis of anthracycline cardiomyopathy is variable and depends on numerous factors, including the age and underlying prechemotherapy cardiac status of the patient and the time of presentation relative to the last dose of drug. Subjects who present late (several months) or very late (years) after the last dose have a better prognosis because the anthracycline myocardial effect takes at least 60 days to become fully manifest. That is, subjects who develop heart failure within a few days of the last dose of drug have an additional cardiomyopathic burden to face, since the last one to two doses produce their full morphologic effect over the next 1 to 2 months.
TREATMENT/PREVENTION
Subjects who develop anthracycline cardiomyopathy should be treated aggressively with conventional heart failure treatment, since some degree of reversibility is likely. Conventional treatment consists of ACE inhibitors, digoxin, and diuretics. Beta-Adrenergic blockade has been used successfully in some subjects, but because of the high adrenergic drive, it may be difficult to administer. On the other hand, the heightened adrenergic mechanism may be producing a commensurate amount of adverse effect on the myocardium, and so the potential for a favorable response may be even greater than in other kinds of cardiomyopathy. In severe refractory cases, cardiac transplantation may be performed provided that the patient's cancer is in complete remission and is not likely to recur (approximately70 percent chance of cure).
Several strategies have been shown to lower the risk of developing
anthracycline cardiomyopathy without compromising the chemotherapy
response rate. These include using endomyocardial biopsy and
right-sided heart catheterization with exercise to assess risk,
which virtually eliminates clinical cardiomyopathy and allows
more anthracycline to be administered to less susceptible subjects;
using serial radionuclide angiography with or without exercise
as a monitoring strategy, which may be somewhat helpful but
because of a low specificity reduces the total amount of chemotherapy
that can be administered safely to some subjects; giving the
agents as low-dose weekly or as 48- to 72-h infusions rather
than as every 3- to 4-week boluses; using a liposomal formulation
; or concomitantly administering a second agent that reduces
toxicity. Unfortunately, none of these strategies completely
eliminates the risk of developing a clinical cardiomyopathy.
PostpartumCardiomyopathy
DEFINITION/DIAGNOSIS
Postpartum or peripartum cardiomyopathy is defined as the presentation
of systolic dysfunction and clinical heart failure during the
last trimester of pregnancy or within 6 months of delivery.
Given the extreme hemodynamic load produced by pregnancy, it
is perhaps surprising that postpartum cardiomyopathy is not
more common.
DISTINCT PATHOPHYSIOLOGY
Postpartum cardiomyopathy most likely will be classified within the "dilated" WHO/ISFC category but occasionally will be "unclassified" because dilatation and remodeling have not had time to occur. Postpartum cardiomyopathy is likely a heterogeneous group of disorders consisting of the addition of the hemodynamic load of pregnancy to a variety of underlying myocardial processes, including hypertensive heart disease, familial or idiopathic
PROGNOSIS
Approximately half of subjects who develop postpartum cardiomyopathy will recover completely, and the majority of the rest will improve. Subjects who have developed a postpartum cardiomyopathy should never become pregnant again, even if myocardial function has recovered fully.
TREATMENT
Treatment should be aggressive and as
for IDC. Cardiac transplantation may be required in severely
compromised patients who do not improve.
N. Friedreich Ataxia
O. Sarcoidosis
SARCOIDOSIS
PATHOGENESIS
Sarcoidosis is a systemic granulomatous disease of unknown etiology characterized by enhanced cellular immune responses. The patholo
gic hallmark of this disease is the noncaseating granuloma (figure 77 c).The initial lesion is an inflammatory infiltrate consisting of activated helper-induced T lymphocytes and abundant macrophages that secrets cytokines. The macrophages aggregates and the differentiate into epitheliod and multinuclear giant cells. Fibroblasts, mast cells, collagen fibers and proteoglycans encase the inflammatory cells into a ball-like cluster. The fibrotic response results in end-organ damage.
Clusters of cases have been observed, suggesting
spread by person to person-to-person exposure or environmental
agents/pathogens.
Genetic factors may also play a role in the development of the
disease as an exaggerated cellular immune response and information
of granulomas may develop in genetically predisposed hosts after
exposure to the offending antigen.
CLINICAL PRESENTATION
The clinical manifestations of sarcoidosis are protean. The disease may be widespread or limited to a single organ. Virtually any organ except the adrenal gland may be involved. The lymphoid , pulmonary, cardiovascular, hepatobiliary, and hematologic systems are the most commonly involved, with the lungs being affected in over 90 percent of patients.
Cardiac sarcoid is more common than previously recognized. In a recent autopsy study of 38 patients with sarcoidosis, 76 percent had cardiac involvement, accounting for 50 percent of the deaths. In other series, sarcoidosis affected the heart in 25 to 50 percent of autopsy cases with fatality in 50 percent of the cases with cardiac involvement. Cardiac sarcoid is more likely fatal and less likely to be diagnosed antemortem than pulmonary sarcoid,. Cardiac sarcoid is more commmonly than previously recognized. In arecent autopsy of 38 patients with sarcoid, 76 % had cardiac involvement accounting for 50% of the deaths. In other series,,sarcoidosisaffeccted the heartt in 25 to 50% of autopsy cases with fatality i 50% of hte cases with cardica involvment.Cardiacx sarcoid is more likey fatal and less likely to be diagnosed antemortem than pulmonary sarcoid.. Frequently the initial presentation is that a sudden death. Myocardial involvement peaks between the third and sixth decades of life. Less than 10 per cent of patients with sarcoid have symptoms referable to the cardiovascular system.
In myocardial sarcoid, portions of the myocardial
wall are replaced by sarcoid granulomas, which preferentially
involve the cephalad portion of the ventricular septum or the
left ventricular papillary muscles. Myocardial involvement is
much more common than pericardial involvement. Cor pulmonale
due extensive pulmonary sarcoidosis with interstitial fibrosis
may occur.
Because of the varied extent of the myocardial granulomas, presenting
signs and symptoms range from first degree heart block to fulminant
heart failure. First degree AV block, bundle -branch block,
complete heart block,ventricular arrhythmias ,sudden death,
and heart failure occur with a frequency of 10 to 20 per cent.
Heart failure can present as s cardiomyopathy with restrictive
hemodynamics.Some 25 percent of the deaths due to cardiac sarcoid
are from heart failure, while sudden death accounts for one-third
to one-half of the deaths.
DIAGNOSIS
In diagnosing cardiac sarcoid, evidence of
other organ system involvement including lymphadenopathy, hepatomegaly,
splenomegaly, or pulmonary findings should be sought. In cases
where the heart is involved to a much greater degree than are
other organs little or no evidence of extra cardiac sarcoidosis
may be found. Chest x-ray, ECG and echocardiography findings
will depend on the extent and location of involvement .
Due to the scattered nature of the granulomas, endomyocardial
biopsy lacks sensitivity and seldom makes the diaognosis despite
high specificity. Magnetic resonance imaging has been useful
in diagnosing scars or lesions in the myocardium due to sarcoid.
TREATMENT
Although no controlled trials have been performed, high dose corticosteroids are usually given in the hope that the course of the disease may be altered. Administration of corticosteroids can improve cardiac symptoms and reverse ECG changes in over half of treated patients. Antiarrhythmic drugs should be used as necessary, although drug therapy of ventricular tachycardia in patients with sarcoidosis, even when guided with program ventricular stimulation, is associated with a high rate of arrhythmia reoccurrence or sudden death. Automatic internal cardioverter-defibrillators have been advocated. Prognosis after the diagnosis of cardiac sarcoid is variable but can the poor. In one series of 247 patients, survival was 41 percent at five years and fifteen percent at ten years. Transplantation is also a successful treatment, as recurrences of sarcoid in the allograft is low, possibly due to post transplants steroid therapy.
P. Hypersensitivity
Q. Noncompaction Syndrome of the Left Ventricle, Endocardial Fibroelastosis(EFE), Barth Syndrome
Noncompaction of the left ventricular
myocardium is characterized by numerous, prominent ventricular
trabeculations, deep intertrabecular recesses (areas of abnormal
blood lakes within the endocardium), arrhythmias and a distinctive
facial dysmorphism. The right ventricle may be similarly involved
with these markedly abnormal trabeculations. There is no abnormal
wall thickening. Wall thickness is normal.
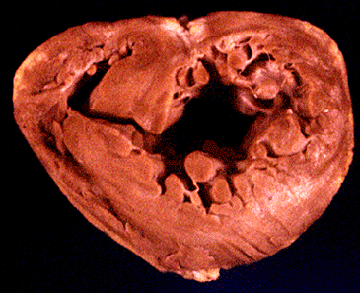
Figure1.
There is endocardial thickening in cases of endocardial fibroelastosis, leading to decreased compliance and impaired diastolic function.Primary forms are typically unassociated with other cardiac anomalies. It usually presents in infancy and early childhood with signs and symptoms of congestive heart failure. The diagnosis is made by biopsy. Treatment with anticongestive and inotropic measures have been ineffective, and the clinical course usually results in death or transplantation. Histopathology reveals extensive deposition of extracellular matrix (collagen and elasticfibers)
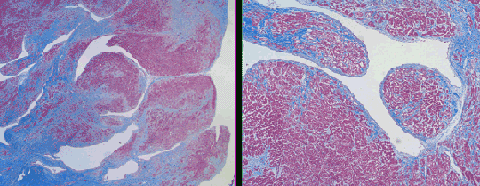
Figure 2.
Three inherited forms have been described. The majority of cases occur sporadically. The X-linked form shows miitochondrial abnormalities similar to Barth syndrome with the exception that EFE patients have endocardial scarring. It is likely that this form is caused by mutations in the G4.5 gene found in Barth syndrome and LV noncompaction.
The incidence of the sporadic form has diminished markedly in the U.S. with the giving of vaccine (mumps-measles-rubella) to many in the population.
The Barth syndrome has skeletal muscle involvement and white cell abnormalities as well as cardiac abnormalities.
But all three have mutations in the G4.5,
as does dilated hypertrophic cardiomyopathy (see above)..
Cardiac arrhythmias including ventricular tachycardia are common in these cases, and hence implantable cardiac defibrillators are advised.
At times, as the problem progresses and cardiac failure occurs,there has been the need to do heart transplants when medication fails.
Non-compaction of Myocardium Cardiomyopathy
Non-compaction of the ventricular myocardium ("spongy myocardium") is a rare congenital cardiomyopathy of children and adults resulting from arrested myocardial development during embryogenesis. Prior to formation of the epicardial coronary circulation at about 8 weeks of life, the myocardium is a meshwork of interwoven myocardial fibers that form trabeculae and deep trabecular recesses. That increased surface area permits perfusion of the myocardium by direct communication with the left ventricular cavity. Normally,as the myocardium undergoes gradual compaction, the epicardial
In this disorder, echocardiography demonstrates a thin epicardium with extremely hypertrophied endocardium and prominent trabeculations with deep recesses. These features tend to be apically localized since compaction would normally proceed from base to apex, and from epicardium to endocardium.
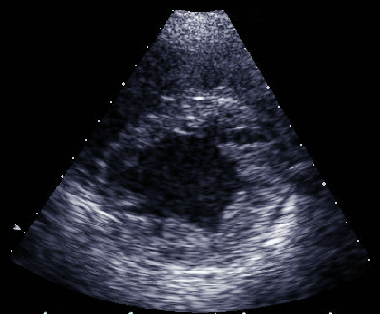
Figure 3.
Clinical presentation consists of congestive heart failure with depressed left ventricular systolic function, ventricular arrhythmias, arterial thromboemboli from thrombus formation within the inter - trabecular recesses, as well as restrictive physiology from endocardial fibrosis.
INCIDENCE AND NATURAL HISTORY OF LEFT VENTRICULAR NON-COMPACTION PRESENTING DURING CHILDHOOD.
P. Daubeney, A. Nugent, P. Chondros, L. Wilkinson, A.M. Davis, S. Kleinert, C.W. Chow, J.L. Wilkinson, R. Weintraub.
Departments of Cardiology, Clinical Epidemiology & Biostatistics, and Anatomic Pathology, Royal Children's
www.csanz.edu.au/abstracts/48abstracts/343.htm
Left-ventricular non-compaction (LVNC) has previously been considered a rare medical curiosity whose cause and outcome are unknown. This review examines the clinical features and outcomes for children with LVNC who were enrolled in the National Australian Childhood Cardiomyopathy Study. This is an ongoing population-based study which includes all children within Australia with primary cardiomyopathy who presented at 0-10 years of age.
The diagnosis was based on the presence of characteristic honeycomb or spongiform appearance of ventricular myocardium on echocardiography, angiography, cardiac MRI or direct examination. The prognostic factors sought included age at presentation, gender, dominant pathophysiology (DCM or RCM), ventricular systolic dysfunction at presentation, number of affected myocardial segments and presence of Barth syndrome. A total of 21 patients were identified, representing 6.5% of the NACCS study population. The diagnosis was made from echocardiography in 21, ventricular angiography in 11, cardiac MRI in 1 and direct examination (transplant/post-mortem) in 4.
The median age at presentation was 0.4 years (range 1 day - 9.7 years). CHF was the presenting symptom in 17/21 (81%) and 14 patients (67%) presented prior to 12 months of age. Barth syndrome was present in 5/21 (24%). The dominant pathophysiology was DCM in 16 (76%), RCM in 4 (19%) with one child (5%) having normal cardiac function.
Survival free from cardiac transplantation was 55% at 10 years of age and 30% after 15 years. Survival free from late cardiac dysfunction (defined by death, transplant, FS<25% or arrhythmias requiring therapy) was 35% at 10 years and 15% at 16 years. Barth syndrome was the only variable related to outcome. Children with Barth syndrome had a superior 10 year survival (100% vs 25%) and freedom from cardiac dysfunction (75% vs 12%; P<.02 for both) compared to the remaining study
We conclude that LVNC is more frequent than previously recognised. Patients without Barth syndrome have a poor prognosis and merit early consideration of cardiac transplantation.