Congestive heart failure is a clinical syndrome resulting from
a primary abnormality in the function of the heart as a pump,
and its diagnosis and therapy depend on an understanding of
the underlying pathophysiology. This pathophysiology involves
a wide variety of initial cardiac events as well as vascular
and neurohumoral compensatory responses that contribute not
only to the clinical syndrome, but also to its progression and
premature mortality.
Primary cardiac overloads (e.g.,
hypertension or valve disease, like mitral insufficiency or
stenosis, aortic stenosis or insufficiency, see definitions
hypertension,
mitral valve,
aortic stenosis,
aortic insufficiency) or loss of myocardium (e.g., myocardial
infarction, heart attack, see definition myocardial
infarction) elicite adaptive myocardial (heart muscle) changes
including hypertrophy (enlargement of muscle cells) and ventricular
chamber dilation. These adaptations may result in myocardial
dysfunction that, if extensive enough, produces ventricular
dysfunction, which ultimately eventuates in the syndrome of
congestive heart failure.
Thus, etiology of the heart disease
forms only one part of the physiologic derangement. The evaluation
and treatment of the overt clinical syndrome involves consideration
of not only the initial cause of the cardiac dysfunction but
also the evolving compensatory process.
Terminology to be used below
1.
Left ventricular dysfunction:
an abnormality of systolic (contraction phase of the heart beat)
or diastolic (relaxation phase when the ventricle refills) performance
of the left ventricle such that either the contractile force
is impaired and ventricular emptying reduced or the diastolic
relaxation and filling are impaired, or both.
a.
Systolic dysfunction:
an impairment of the contraction of the left ventricle such
that the stroke volume (SV, the amount of blood pumped out per
heart beat) is reduced for any given end-diastolic volume (amount
of blood remaining after contraction) or filling pressure. In
general, the end-diastolic volume is increased significantly
while the stroke volume is reduced. This may involve either
or both ventricles.
b.
Diastolic dysfunction: a state in which ventricular
filling rate and the extent of filling is associated with an
inappropriate rise in ventricular diastolic pressure (pressure
after contraction ends). Normal systolic emptying (EF or ejection
fraction)) may be maintained.
2.
Heart failure: a clinical syndrome in which symptoms
are associated with abnormalities in systolic or diastolic function.
These symptoms may include fatigue, exercise limitation, dyspnea
(shortness of breath) on exertion, orthopnea (shortness of breath
relieved by sitting up), or paroxysmal nocturnal dyspnea(sudden
shortness of breath at night).
3.
Congestive heart failure: a clinical
syndrome in which symptoms of cardiac dysfunction as noted under
heart failure are accompanied by signs or symptoms of congestion,
including peripheral edema (ankle swelling) or pulmonary congestion
(rales in the lungs).
4.
Impedance:
the total force opposing left ventricular ejection
of blood out through the aortic valve and composed primarily
of arterial wall compliance, arteriolar resistance, and inertance.
5. Systemic
vascular resistance: the calculation resulting from the
division of the mean systemic arterial pressure, minus the mean
systemic venous pressure, by the cardiac output (amount of blood
pumped out per minute) and representing the sum of the resistance
to flow of all parallel vascular channels in the systemic circulatory
system.
6. Ventricle
remodeling: an adaptive process which the ventricle is
reshaped by structural changes resulting in increased chamber
volume and increased myocardial mass. This involves myocyte
(heart muscle cell) hypertrophy with cell widening and elongation
as well as myocyte rearrangements in the ventricular wall. Interstitial
growth (in between the myocytes) also is characteristic of the
process.
Etiology of Heart Failure
In general, heart failure begins
with either abnormality of coronary blood flow (ischemia and
infarction, see definition of
coronary atherosclerosis, angina and myocardial
infarction in this web site), ventricular overloads whether
from pressure (systemic arterial hypertension, aortic stenosis)
or volume (mitral regurgitation or congenital shunt lesions,
see definition congenital
heart disease), or unexplained myocyte loss and depression
(cardiomyopathies, viral myocarditis, toxins; see definition
cardiomyopathies).
It may also evolve from incessant arrhythmias (sustained tachycardia).
Less commonly, the syndrome of congestive failure can result
from external factors that limit ventricular filling (constrictive
pericarditis). See definitions respectively on this web site.
Ischemic Heart Disease
Both systolic and diastolic dysfunction
are early manifestations of myocardial ischemia, since even
modest reductions of blood flow may deprive the myocardium of
adequate nutrition for generation of muscle contraction and
muscle relaxation.
In humans the induction of ischemia,
through either decreased coronary blood flow or increased oxygen
need not met by increased blood flow, results in a rapid loss
of contraction in the region involved. If blood flow is restored
to the region before necrosis ensues, contractile activity does
not return for hours or even days.
This persistent loss of contractile
activity for hours or days despite return of blood flow and
the absence of necrosis is termed "stunning" and may contribute
to ventricular dysfunction and symptoms of heart failure. Severe
stunning may also produce persistent segmental dilatation of
the ventricle by damaging connective tissue connections between
cells, a form of remodeling.
Another concept, "hibernating"
myocardium, refers to myocardium with a contractile dysfunction
resulting from a chronic inadequacy of blood flow without histologic
evidence of myocardial infarction (see magnetic
resonance definition on this web site, and figures
122,
123,
124,
125).
Myocardial infarction results
in a loss of functioning myocardium in the region served by
the occluded coronary vessel. If the loss of coronary flow is
incomplete perhaps due to collateral blood flow, only a subendocaridal
infarction (occurring just under the inner lining of the ventricle
and not extending through the entire wall to the outer surface
of the heart) may result. Nevertheless, this leaves a larger
load for the myocardium remaining in the region.
The scar resulting from myocardial
infarction may further contribute to left ventricular dysfunction
by restricting filling or by creating an aneurysm (see definition
myocardial
infarction and aneursym
on this web site). With large infarctions, compensatory ventricular
dilation occurs and reactive hypertrophy occurs in the remaining
well-perfused myocardium. With large increases in diastolic
filling pressure, "myocyte slippage" occurs and leads to further
loads on the ventricular wall. These compensation processes
of myocyte hypertrophy, dilatation, and changes in wall conformation
comprise ventricular remodeling.
Mitral regurgitation may also
develop, secondary to either left ventricular dilatation or
papillary muscle (cardiac muscle bundles, which connect to the
mitral or tricuspid valve through chordae tendonae) dysfunction
(see definition on this website, figure
104b).
Thus, although the regional dysfunction
or scar formation association with coronary artery disease initially
is confined to an area in the distribution of the involved coronary
artery, this process often progresses to a dilated ventricle
with global impairment of contractile function (hypokinesis).
This chamber enlargement, or ventricular remodeling, may occur
over days, weeks, months, or years and eventuate in a marked
impairment of systolic function. In other instances, however,
the regional dysfunction may persist and the ventricle may remain
normal in size for many years.
Hypertrophy is an almost invariable
accompaniment of the remodeling process in patients with primary
ischemic disease of the myocardium. This hypertrophy may, in
part, represent a reactive process to normalize wall stress
induced by dilatation of the chamber dimension. Neurohumoral
factors may also contribute to the ventricular hypertrophy.
With these considerations in
mind, therapy of ischemic processes is directed both to the
ischemic event as well as to the ventricular remodeling it induces.
Nonischemic Disease
In a patient with epicardial
coronary arteries that angiographically appear to be normal
or nearly normal and with no other apparent disease, a dilated,
poorly contracting left ventricle is usually diagnosed as idiopathic
dilated cardiomyopathy (figure
43b). Nonetheless, even in this clinical situation ischemia
cannot be excluded as an important etiologic factor. Small vessel
disease that can influence regional or global perfusion may
not be demonstrable by angiographic techniques.
A primary disease affecting
the myocyte and its contractile process appears to be the cause
of most cases of idiopathic dilated cardiomyopathy (see Definition
Cardiomyopathy on this web site, and figure126).
In general, idiopathic dilated cardiomyopathy is characterized
by focal, diffuse myocyte loss, replacement fibrosis (scar like
tissue), and reactive hypertrophy of the remaining myocytes.
A variety of etiologic agents
has been implicated, and the process can be multifactorial.
In most instances the etiology of cardiomyopathy or heart muscle
disease of unknown cause cannot be determined by histological
examination. Aside from systemic arterial hypertension and coronary
artery atherosclerosis, viruses, alcohol, and diabetes may represent
the most prevalent factors contributing to heart muscle dysfunction
in North America. Diabetes may also be associated with large
and small vessel obstructive disease.
Due to increasingly effective
therapy for hypertension, the incidence of hypertension as a
primary cause of heart failure has been greatly reduced, but
remains an important cofactor in heart failure from other etiologies.
PATHOPHYSIOLOGY
Ventricular Function
The myocardial abnormalities
described above ultimately result in hemodynamic derangement
of ventricular function. As capacity for force development and
shortening by the ventricular wall is lost, a compensatory increase
in diastolic ventricular volume occurs. Thus, stroke volume
is only maintained by an increase in diastolic volume and pressure.
Moreover, the relation of stroke
volume to end-diastolic pressure is not only depressed but flattened
at higher filling pressure. Should marked ventricular hypertrophy
without myocyte loss occur, such as with systemic arterial hypertension,
prolonged systole, and delayed or incomplete diastolic relaxation
of the ventricle, along with thicker ventricular wall, may elevate
the end-diastolic filling pressures even with normal volumes,
and the end-diastolic pressure be actually less than expected
for the increase in volume.
In the early stages of left ventricular
dysfunction, the hemodynamic abnormalities may be confined to
exercise, when the increase in stroke volume demanded by exercise
may be inadequate and/or accompanied by an abnormally brisk
increase in left ventricular filling pressure. The left ventricle
normally is capable of adjusting its work output to match an
increasing aortic impedance over a wide physiological range.
The dysfunctional left ventricle
loses this ability and its performance becomes progressively
more impaired as the impedance is increased. Thus, the failing
left ventricle becomes very sensitive to impedance or afterload
and, becomes quite insensitive to preload. This important physiological
shift from a preload-dependent and afterload-independent ventricle
in the normal individual to a preload-independent and afterload-dependent
ventricle in the setting of heart failure has important therapeutic
implications.
In most patients with heart failure
some degree of diastolic dysfunction accompanies systolic dysfunction.
The left ventricular diastolic dysfunction, or reduced diastolic
ventricular compliance, implies that the left ventricular is
stiffer than normal and therefore responds to a small increment
in volume with a prominent increase in diastolic filling pressure
that is transmitted backward into the pulmonary vasculature,
leading ultimately to pulmonary congestion rales,and dyspnea..
Neurohormonal Activation
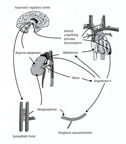
Neurohormonal systems are activated
in the setting of left ventricular dysfunction (regardless of
etiology) with decreased cardiac output and consequently arterial
underfilling and activation of baroreceptors in the aortic arch,
carotid sinus, and left ventricle (figure151).
These baroreceptors stimulate vasomotor regulatory centers in
the medulla, which activate the sympathetic nervous system(see
definition autonomic nervous system), arginine-vasopressin system,
and renin-angiotensin-aldosterone system.
Activation of the sympathetic
nervous system is manifested by elevated plasma norepinephrine
levels, increased spillover into the bloodstream of norepinephrine
released into the synaptic cleft, and evidence for increased
sympathetic nerve traffic (increases in heart rate, myocardial
contractility, peripheral vasoconstriction).
The renin-angiotensin system
activated in heart failure, presumably through intrarenal mechanisms
stimulated by changes either in pressure or changes in sodium
load in the macula densa, leads to sodium and water retention.
Peripheral Vasculature
An increase in systemic vascular
resistance and a reduction of arterial and venous compliance
are hallmarks of the syndrome of heart failure.
Clinical Manifestation of Heart Failure
The four major clinical manifestations
of heart failure are left ventricular dysfunction, exercise
intolerance, congestion or edema, and ventricular arrhythmias.
Chronic heart failure symptoms
can be divided into four classes according to the New York Heart
Association (NYHA) as follows:
Class1:
Symptoms cause no limitation of physical activity. Ordinary
physical activity does not lead to undue fatigue, or dyspnea.
Class2:
Symptoms cause slight limitation of physical activity.Patient
is comfortable at rest, but ordinary physical activity results
in fatigue, palpitations,or dyspnea.
Class3:
Symptoms cause marked limitation of physical activity. Patient
is comfortable at rest, but even slight physical activity causes
fatigue ,palpitations, or dyspnea.
Class4:
Symptoms of cardiac insufficiency are present at rest, and discomfort
is increased with any physical activity.
Clinical Diagnosis (see other
related sections on this website for history, physical diagnosis,
and laboratory tests)
1.
The first responsibility of treatment is to correct or stabilize
any primary abnormality or overload that can be identified.
Thus, ischemia is controlled by medical or surgical intervention,
hypertension is rigorously treated, and primary valve abnormalities
are evaluated for the possibility of repair. ( see other related
sections of this website)
2. Nonpharmacologic Therapy
A.
Salt restriction B. Weight loss
C. Restriction of dietary saturated
fat and cholesterol D. No smoking
E. Exercise prescription
3. Pharmacologic Therapy
A.
Diuretics
B. Vasodilator Drugs (to relax the
systemic vasculature and/or reduce venous tone)
i. Nitrate (isosorbide)
ii. Hydralazine (Especially when added to a regimen of digoxin
and diuretic therapy)
iii. Ace inhibitors (captopril, enalapril)
C. Inotropic Drugs (positive ones
which increase the contractile force of the myocardium)
i. digitalis glycosides (i.e.digoxin)
D. Neurohormonal Inhibition
i. Ace inhibitors inhibit the conversion
of angiotensin1 to angiotensin 2(A2) through angiotensin-converting
enzyme(ACE) causing a reduction in circulating and tissue levels
of angiotensin11 and a reduction in norepinephrine (through
down-regulation of the sympathetic nervous system see definition
autonomic
nervous system), subsequent reduction in aldosterone leading
to decrease in sodium and fluid retention, possble inhibition
of myocardial hypertrophy and remodeling and vascular hypertrophy,
reduction of resistance to left ventricular outflow through
vasodilation.
A recent study has shown that
enalapril therapy (an angiotensin-converting-enzyme inhibitor)
is associated with a significant reduction in the risk of hospitalizationfor
heart failure among white patients with left ventricular dysfunction,
but not not among similar black patients. This difference may
be related to the lower plasma renin levels and endogenous nitric
acid levels in black patients as well as to under lying genetic
determinants of drug response.
Reference: Exner,D.V.,et. als.,Lesser Response
To Angiotensin-Converting-Enzyme Inhibitor Therapy In Blcak
As Compared With White Patients With Left Ventricular Dysfunction,N.Engl.J.Med.,Vol.344,No.18,May3,20011351-57.
ii.
Ace2 receptor blockers (ie, losartan) block A2 from all pathways
at the receptor level, producing vasodilation and inhibiting
muscle cell proliferation. But these agents are to be used only
if the ACE inhibitors can not be tolerated due to side effects
such as cough, since a recent study showed no statistical difference
in mortality rates between the captopril and the losartan groups.
iii. Beta blockers improve left
ventricular function,symptoms and functional class, and prolong
survival in patients with CHF due to left ventricular dysfunction.
Currently, carvedilol,metoprolol,and
bisoprolol are proven therapies. Carvedilol in addition has
alpha-receptor-blocking effects, which theoretically gives more
complete sympathetic blockade.
It has been shown that one year
of treatment with carvedilol in dialysis patients with congestive
heart failure (CHF) and dilated cardiomyocardiopathy (DCM) reduced
LV volumes and improved LV function and clinical status.
Beta blockers modify the dysregulated
cytokine network (interleukin-10, tumor necrosis factor-alpha
(TNF-alpha), and soluble TNF receptors (sTNF-R-1 and R2) in
dilated cardiomyopathy and may be responsible for the efficacy
of therapuetic drugs for heart failure.
A recent study showed that the
benefit of carvedilol was apparent and of similar magnitude
in both black and nonblack patients with heart failure.
Reference:Yancy,C.W.,Et.Als.Race and the Response
to Adrenergic Blockage with Carvedilol in Patients with Chronic
Heart Failure,N.Engl.J.Med.,Vol.344.No.18,May 3,2001,1358-65.
Also, another recently reported
study reveals that carvedilol's benefit extends to patients
in severe heart failure with regards to morbidity and mortality.
Reference:Packer,M. and others,Effect of Carvedilol
on Survival in Severe Chronic Heart Failure,N.Engl.J.Med.,Vol.344,No.22,May31,2001,1651-1658.
On the other hand, another study
published in the same journal at the same time indicate that
in a group of patients with NYHA class111 and 1V treatment with
bucindolol(a beta-blocker) resulted in no significant overall
survival benefit. But there was a benefit in nonblack patients.
One possible reason for the difference
between the two preceding studies may be due to the different
pharmacologic properties of bucindolol, a nonselective beta-blocking
agent with unique sympatholytic activity due to its strong B2-adrenergic
blockade and only weak alpha-1-blocking properties.
Reference:The Bata-Blocker Evaluation of Survival
Trial Investigators, A Trial of the Beta-Blocker Bucindolol
in Patients with Advanced Chronic Heart Failure,N.Engl.J.Med.,Vol.344,No.22,May31,2001,1659-1667.
But it has been pointed out that
the most advanced forms of heart failure (not extremely severe)
patients were excluded in the first referred to study above
and an improvement was observed in nonblack patients. It is
important to remember that these drugs must be administered
and the dose escalated slowly in patients with heart failure,
especially those in whom the condition is severe.
Indications for beta-blockers:
1.
Treat all patients who have NYHA class2 or 3 symptoms.
2.
Consider therapy for patients with NYHA class1 or 4 symptoms.
3.
Use agents that have proved beneficial in major mortality trials
(see above second paragraph).
4.
Before adding beta- blocker therapy, make sure patient is stable
and on standard heart failure treatment (eg, ACE inhibitor,
diuretic, digoxin).
5.
Start beta-blocker theray at low dosage (eg,carvedilol, 3.125
mg PO bid; metoprolol CR/XL, 12.5 mg PO qd; bisoprolol, 1.25
mg PO qd)
6.
Increase dosages at 2 to 3 wk intervals, as tolerated to target
levels established in major mortality trails (eg, carvedilol,
25-50 mg PO bid; metoprolol CR/XL, 200 mg PO qd; bisoprolol,
10 mg PO qd)
7.
Tell Patients, Side effects may occur early in therapy but do
not preclude long-term use. Symptomatic improvement may not
be noticed for 2 to 3 months. Treatment may reduce risk of disease
progression, even if symptoms do not improve.
Contraindications to beta-blocker
therapy for CHF: signs of clinically unstable heart failure
in the previous 2 weeks: a). increase
in body weight, b). increase in
diuretic dose, c). need for intravenous
diuretic ot inotrpic ageents, d).
need for hospitalization for cardiac symptoms, 4).
presence of episodic worsening of CHF symptoms, e).
bronchial asthma or emphysema sensitive to beta agonists, f).
bradycardia (heart rate<60beats perminute, g).
hypotension (systolic blood pressure<100mmHg), h).
second or third degree heart block.
iv.
Aldosterone antagonists such as spironolactone should be considered
in all patients with severe symptomatic heart failure in the
absence of significant renal insufficiency or hyperkalemia.
v.
Preliminary data suggest that endothelin receptor antagonists,
vasopeptidase inhibitors, and synthetic natriuretic peptides
may represent the next wave of CHF treatment options.
Reference:Cice,Gennaro,MD
and others.Dilated Cardiomyopathy in Dialysis Patient-Beneficial
Effects of Carvedilol:A Double-Blind, Placebo-Controlled,Trial,Jacc,,vol.37,no2,2001
February 2001:398-406
Reference:Packer,M.
and others,for the US Carvediol Heart FAILURE STUDY STUDY GROUP.The
effect of carvedilol on morbidity and mortality in patients
with chronic heart failure,NEJM 1996;334:1349-55.
Ohtsuzki,T.
MD,and others,Effect of Beta-blockers on Circulating Levels
of Inflammatory and Antiflammatory Cytokines in Patients with
Dilated Cardoimyopathy,JACC,VOL37,nO.2,2001 PP.412-417.
Ward,R.P.
and Anderson,A.S.,Slowing the progression of CHF,Postgraduate
M ed.,Vol.109,No.3,March2001,36-45.
Garg, Ravi
M.D. and Sorrentino, Matthew MD, Postgraduate Medicine Vol 109/
No 3./ March 2001 pg 49-56
E.
Antiarrhythmic Therapy (may shorten life expectancy and hence
are used with caution during symptomatic ventricular arrhythmias
in view of the risk. In such patients other pharmacologic agents
(? like amiodarone) or implantible defibrillators may be more
effective therapy. See definition ventricular
tachycardia on this web site).
F.
Anticoagulant Therapy (to reduce the risk of systemic embolization
in patients with atrial fibrillation, but it is not indicated
in patients who are fairly active and have not had a previous
episode of embolization).
G.
Biventricular Pacing in Patients with Severe Congestive Heart
Failure due to Idiopahitic or Ischemic Heart Left Ventricular
Systolic Dysfunction with Intraventricular conduction delay
(QRS complex duration, see figure
94 re normal EKG) of more than 150ms. (over 100ms is abnormal)
and without a standard indication for insertion of a pacemaker
(see definition bradycardia
management).
This procedure of atriobiventricular
pacing has been shown to significantly improve exercise tolerance,
symptoms, and the quality of life in these patients and is associated
with a reduced number of hospitalizations for decompensated
heart failure.
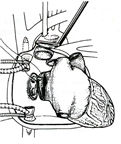
It involves the implantation
of all leads transvenously. The atrial lead is placed high in
the right atrium. The left ventricular lead is placed in a tributary
of the coronary sinus, according to a previously described method
(figure
149). Specially designed electrodes are used. A venogram
helps to optimize the position of the leads (figure 150). The
target area was preferably the lateral wall, midway between
base and apex, but other lateral or posterior sites were also
acceptable. The great cardiac vein or the mid cardiac is used
only when other sites were not accessible. The right ventricular
lead was positioned as far as possible from the the left ventricular
lead.
The pacemakers were triple-output
devices that made use of standard dual-chamber technology, with
built-in adapters to synchronize the pacing of the two ventricles.
(Chorum
7336MSP,ELA Medical,Montroube,France, and InSync 8040,Medtronic,
Minneapolis).
Reference:Cazeau,S. and Others,Effects of Multisite Biventricular
Pacing in Patients with Heart Failure and Intraventricular Conduction
Delay,NEJ of M.ed.,March22,2001,No.12
V.344.pp873-880.
Reference:Dauber,J.C.,and Others,Permanent Left Ventricular
Pacing With Transvenous Leads Inserted Into The Coronary Veins,Pacing
Clin.Electrophysiol.1998;21;239-245
4. Invasive
Approaches
A. Coronary Reperfusion,especially
if there is the occurence of repeated episodes of acute left
heart failure associated with pulmonary edema even in the absence
of chest pain and dietary salt indiscretions (suggesting "flash"
or acute ischemic left ventricular diastolic dysfunction. Coronary
angography may be indicated. See coronary angiography definition
on this web site).
If ischemia is obvious, the presence
of left ventricular dysfunction is not a deterrent to coronary
revascularization.
B. Valvular Heart Disease (surgery
for functional valvular regurgitation due to left ventricular
heart failure is not very effective and very risky).
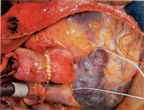
C.
Reduction ventriculoplasty involves excising the part of the
left ventricular muscle which is dyskinetic, resulting in an
increase in the contracting myocardium that maintains cardiac
output (figure
151b). It offers a potential solution to a subset of patients
with end stage heart failure.
D.
Transmyocardial laser revascularization may be used in patients
deemed ineligible for bypass surgery or any percutaneous intervention.
It involves using a laser instrument to drill holes into the
myocardium, which sets up an inflammatory reaction around the
area, to neovascularization to increase the blood supply to
the ischemic area.
E.
Procedures to assist or replace heart function:
1) intra-aortic balloon pump (figure 152),
2) permanent implantable balloon pump (figure 153),
3) total artificial heart (figure 154).
F.
Heart Transplantation (most effective therapy for severe heart
failure, figure 155).
Reference:Cohen,J. and
Sonnenblick,E.H., Diagnosis and Therapy of Heart Failure, Hurst's
THE HEART,8th Edition ,pp.557-571
Reference:Jayakar,D.V.,Surgical
treatment of CHF,Postgraduate Med. vol.109/n.3,March2001,61-69
RIGHT VENTRICULAR HEART
FALURE
OBSTRUCTIVE SLEEP APNEA,
HYPOVENTILATION, PULMONARY HYPERTENSION, RIGHT VENTRICULAR FAILURE
There are numerous respiratory
complications of obesity, including an increased breathing workload,respiratory
muscle inefficiency,decreased functional reserve capacity and
expiratory reserve volume ,and closure of peripheral lung units.These
complications often result in a ventilation-perfusion mismatch,especially
in the supine position.Obesity is a classic
cause of alveolar hypoventilation. Historically,
the obesity-hypoventilation syndrome has been described as the
"pickwickian" syndrome, and obstructive apnea was
first observed in patients with severe obesity. Sleep apnea
is defined as repeated episodes of obstructive apnea and hypopnea
during sleep, together with daytime somnolence and/or altered
cardiopulmonary function. The prevalence of sleep-disordered
breathing and sleep disturbances rises dramatically in obese
subjects and obesity is by far the most important modifiable
risk factor in sleep-disordered breathing.It has been estimated
that 40 million Americans suffer from sleep disorders and that
the vast majority of these patients remain
undiagnosed.
Despite careful screening by history and physical examination,
sleep apnea is revealed only by polysomnography in most patients.
Although, some clinical features could be useful in screening
for sleep apnea, the diagnostic accuracy is inadequate.
Patients with sleep apnea have an increased risk of diurnal
hypertension, nocturnal dysrhythmias, pulmonary hypertension,
right and left ventricular failure, myocardial infarction, stroke,
and mortality. The prevalence of pulmonary hypertension in subjects
with obstructive sleep apnea is 15 to 20 percent; however, pulmonary
hypertension rarely is observed in the absence of daytime hypoxemia.
According to Kessler and colleagues the extent of pulmonary
hypertension in patients with obstructive sleep apnea is generally
mild to moderate (pulmonary artery pressures ranging between
20 and 35 mmHg) and does not necessitate specific treatment.
Although there is a link between sleep apnea and systemic hypertension,
the association of obesity with both disorders confounds the
relationship. A physician who evaluates an obese patient who
has been referred for hypertension should address related symptoms
such as habitual snoring, nocturnal gasping or choking, witnessed
episodes of apnea, and daytime sleepiness. It is important to
remember, however, that the clinical and ECG signs of cor pulmonale
appear later than do those of pulmonary hypertension (assessed
by right heart catheterization). Numerous treatments are available
for sleep apnea, but weight loss in obese patients should always
be advocated.
Severe untreated sleep-disordered breathing
can further impair LV function,leading to arterial oxyhemoglobin
desaturation and arrhythmias.Central sleep apnea may occur in
as many as 40% of patients with heart failure,and 10% suffer
from obstructive sleep apnea.Obstructive sleep apnea increases
afterload and heart rate during sleep but is responsive to continuous
positive airway pressure.